


1. Introduction
Loratadine is a second-generation H1 histamine antagonist drug used to treat allergies [1], it is closely related to tricyclic antidepressants [2]. In studies exploring physiological indices of CNS functioning, such as EEG-evoked potentials, and sleep latency tests, loratadine has been shown to be free of CNS effects [3].
Loratadine is available as a generic drug and is marketed for its nonsedating properties and is indicated for the relief of nasal and nonnasal symptoms of seasonal allergic rhinitis and for the treatment of chronic idiopathic urticaria [4].
Besides, loratadine is available in the market in several dosage forms, namely, tablets, syrup, and rapidly desintigrating tablets [5]. It is contraindicated in patients who are hypersensitive to this medication or to any of its ingredients. Patients with liver impairment or renal insufficiency (GFR < 30 mL/min) should be given a lower initial dose (10 mg every other day) [6].
Oral disintegrating tablets (ODT) is defined as a solid dosage form containing medicinal substances, which disintegrates rapidly, usually within a matter of seconds, when placed upon the tongue [7] The significance of these dosage forms is highlighted by the adoption of the term, “Orodispersible Tablet”, by the European Pharmacopoeia which describes it as a tablet that can be placed in oral cavity where it disperses rapidly before swallowing [8].
The most frequent symptoms for respiratory conditions are throat soreness and cough; therefore, a new formulation combining loratadine and ambroxol hydrochloride was designed to treat these 2 major symptoms [9].
It worthy to mention that, different analytical methods used for determination of loratadine in biological samples using HPLC-UV methods shows poor quantitation limits LLOQ above 33ng/ml [10]. Analytical method using HPLC coupled with fluorescence detector showed enhanced sensitivery with LLOQ of 0.5ng/ml after extraction of loratadine with organic solvent followed by back extraction with diluted phosphoric acid [11].
Moreover, for more sensitive determination, LC/MS/MS is used for determination of loratadine in human plasma by protein precipitation of human plasma samples , the detection of loratadine and internal standard was in MRM mode using an ion trap mass spectrometer with electrospray positive ionisation. The ion transitions monitored were 383→337 for loratadine, and 300→226.8 for internal standard [12].
Besides, a sensitive and selective liquid chromatography coupled with atmospheric pressure chemical ionization-mass spectrometry (APCI-LC-MS) method for quantification of loratadine (L) and its active metabolite descarboethoxyloratadine (DCL) in human serum was validated. The detection with m/z 383 for L, and 311 for DCL. The limits of quantification were established at 0.1 ng/mL of L and 0.2 ng/mL of DCL, respectively, with an accuracy and precision less than 9%. Recoveries of both analytes were around 80% [13].
For the best sensitive assay and better sample clean up, attaining quantitation limit of 10pg/ml.The tandem mass spectrometric detection was by monitoring singly charged precursor product ion transitions: 383→337(m/z) for loratadine, 388→342(m/z) for LOR-d(3). The low limit of quantitation (LLOQ) was 10 pg/ml [14].
This study was performed to investigate the bioequivalence of loratadine between a generic product of 10mg oral disintegrated Tablets and Reference Product. The study protocol called for 24 healthy volunteers [15]. The bioanalysis of clinical plasma samples was accomplished through development of an LC/MS/MS method, which was validated in accordance with the international guidelines [16]. Pharmcokinetic parameters, determined by standard noncompartmental methods, and analysis of variance (ANOVA) statistics was calculated. The significance of a sequence effect was tested using the subjects nested in sequence as the error term. The 90% confidence intervals for the ratio (or difference) between the test and reference product pharmacokinetic parameters of AUC0-t and AUC0-inf and Cmax were calculated [17].
2. Materials and Methods
2.1 Chemicals and reagents
- Water of HPLC grade.
- Human plasma (Vacsera Blood Bank).
- Methanol HPLC Grade (Scharlau, spain).
- Acetonitrile of HPLC Grade (Scharlau, spain).
- Diethyl ether (MTEDA, U.S.A.).
- Dichloromethane. (MTEDA, U.S.A.).
- Sodium Hydroxide (Scharlau, spain).
- Formic acid of Reagent Grade (SIGMA-ALDRICH, Germany).
2.2 Equipment
- Adjustable Pippettes (P200, P1000 and P10000).
- Disposable plastic pipettes tips - Labtip Yellow (range 5 - 200 µL) & Labtip Blue (range 200 1000µL).
- Disposable Glass test tubes 120 x 12 mm.
- Vortex mixer (Boeco, Germany).
- Vacuum pump (Boeco, Germany).
- PH-meters (Boeco, Germany).
- Water purifier (Purelab option- R7ELGA, U. K.).
- Sonicator (Crest, U.S.A.).
- Analytical balance (Sartorius, U.S.A.).
- Concentrator Plus/Vacufuge® plus (Eppendorf, Germany).
2.3 LC-MS/MS Components
- Quaternary pump: Agilent 1200 series, U.S.A.
- Degasser: Agilent 1200 series, U.S.A.
- Autosampler: Agilent 1200 series, U.S.A.
- Mass Detctor: Agilent 6410B Triple Quad, U.S.A.
- Data system: Mass Hunter, Agilent.
- A C8 reversed phase column, Thermo Hypersil Gold C8, 4.6 x 50 mm I.D, 5.0 micron.
- Guard column Thermo hypersil Gold C8, 4 x 3 mm I.D.
3. Methods
3.1 LC/MS/MS assay
Chromatographic conditions: In house developed chromatographic conditions was used. Mobile phase composition is 0.5% formic acid: Acetonitrile (10:90 V/V). The flow rate was set at 0.6ml/min. Injection volume was set at 2 ul. MS/MS 6410B detector was operated at ESI positive mode, m/z was 383?337 for Loratadine, and 705.2?392.2 (Internal standard) Itraconazole. Fragmentor energy was set at 135 and 90 for Loratadine, and (internal standard) Itraconazole. Collision energy was set at 20 for both Loratadine, and internal standard.
Preparation of stock and working solutions: Accurately weigh 10mg of LORATADINE standard. Transfer into a 100ml volumetric flask. Add about 80ml Methanol. Sonicate for 10 minutes. Complete to volume with Methanol. This solution contains 100ug/ml LORATADINE "Solution A". Transfer quantitevly 0.1ml of Solution A into a 100ml volumetric flask and complete to volume with water to obtain 100ng/ml "Solution B".
From "Solution B" the following were prepared:
| Master Solution used | Mililitres taken | Final concentration obtained (ng/ml) | Final volume (ml) |
| "Solution B" | 0.05 | 0.5 | 10 |
| "Solution B" | 0.1 | 1 | 10 |
| "Solution B" | 0.2 | 2 | 10 |
| "Solution B" | 0.4 | 4 | 10 |
| "Solution B" | 0.5 | 5 | 10 |
| "Solution B" | 2 | 20 | 10 |
| "Solution B" | 4 | 40 | 10 |
Itraconazole (Internal Standard) standard solution: Accurately weigh 10mg of Itraconazole Standard. Transfer into a 100ml volumetric flask. Add about 80ml of Methanol. Sonicate for 10 minutes. Complete to volume with Methanol. This solution contains 100ug/ml Itraconazole. Transfer quantitevly 2ml of prepared solution into a 100ml volumetric flask and complete to volume with water to obtain 2000ng/ml Itraconazole.
Preparation of Loratadine Standard concentrations in human plasma: The standard samples in human plasma were prepared by transferring a 50 ul aliquot of the working standard solutions of loratadine at concntrations ranging from 0.5 to 100 ng/ml to a centrifuge tubes containing 0.5 ml of blank human plasma.
Sample preparation: Volunteers human plasma samples, standard samples (500ul) were transferred into appropriate centrifuge test tubes 50ul of the internal standard (Itraconazole working solution 2000ng/ ml) were added. Then samples were Vortex-mixed for approximately 1 minute. 50ul of 0.25M (NaOH) were added. Then samples were Vortex-mixed for approximately 1 minute. 3ml of [Diethyl ether: Dichloromethane (70:30) v/v] were added and Vortex-mix was done for approximately 1 to 2 minutes. Centrifugation of samples was done at 4000rpm for 5 minutes; clear organic supernatant layer were transferred to clean test tube and evaporated till dryness. The remaining residue was reconstituted with 200ul mobile phase and transfered to insert vial for injection and quantitation on LC/MS/MS.
Quantitation: The unkown sample concentration of volunteer human plasma was calculated from the following formula: Y = aX + b. Where Y is the peak area ratio, X is the unkown Concentration of loratadine in human plasma samples, a is the slope of the calibration curve, b is the Y-Intercept.
All the previously mentioned procedure was performed stepwise by means of LC-MS/MS instrument using Mass Hunter Quantitative Analysis software, where it substitutes the obtained peak area ratio in the equation and calculates the concentration which is added in the printed output of the LC-MS/MS device for each chromatogram.
3.2 Bioequivalence study
Subjects: Twenty-four healthy adult, male volunteers participated in this study were subjected to complete physical examination and neurological assessment, urine analysis and blood (hematology, biochemistry, and serology). None of the volunteers had any history of drug or alcohol abuse, nor did they have any acute or chronic gastrointestinal, cardiac, vascular, hepatic, or renal disease. No concurrent medication was allowed during the course of the study. Subjects did not receive any meals for four hours after study dose administration, neither any bevrages drink, nor coffee or tea. At 1:00 p.m. they received a standard meal, and at 6:00 p.m. another meal. The written informed consent for the intended study were reviewed, discussed and then signed by the participant and clinical investigator before the beginning of screening procedure without any obligation on the volunteers to continue if they didn’t want to.
Study design: This study was a single-center, open-label, randomized, single-dose study with Two-way crossover design to compare the bioavailability of loratadine between two products, in 24 healthy adult, male volunteers under fasting conitions with a washout period fourteen days between dosing. The number and disposition of the blood collections as well as the washout period were designed with respect to pharmacokinetic parameters of loratadine.
Sample collection: The number of blood collections for drug analysis was 17 samples in each study period. The volume of blood taken for the determination of Loratadine in plasma was 5ml per sample. The following blood samples for the analysis of Loratadine in plasma were collected at the following intervals: 0 (directly prior to dosing), 10min, 20min, 30min, 45min, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 9.0, 12.0, 24.0, 48.0 and 72.0 hours after the administration. Blood samples were collected into tubes containing EDTA disodium as an anticoagulant slightly shaken, and centrifuged at approximately 4000 r.p.m. for 10 minutes. After centrifugation, plasma samples were transferred directly into a 5ml-plastic tube. These samples were immediately stored at the study site in a freezer at a nominal temperature -80oC until analysis. The label of the collecting tubes had the study’s code number, subject number, study period, and the designated sample number. The total amount of blood loss during the whole study did not exceed 170ml.
Analysis of plasma samples: A high-performance liquid chromatography coupled with Triple Quad Mass Detector LC-MS/ MS was used for the determination of loratadine in human plasma. Samples from 24 subjects (who completed both periods of the study) were analyzed. The bioanalytical method was validated according to the international guidelines.
Pharmacokinetic calculations: The following Pharmacokinetic parameters (variables) of Loratadine were assessed; Maximum plasma concentration (Cmax), Time point of maximum plasma concentration (Tmax), Half-life of drug elimination during the terminal phase (t1/2e), and terminal rate of elimination (Ke), area under plasma concentration-time curve from zero to the last quantifiable concentration estimate (AUC0-t), Mean Residence Time (MRT), Area under plasma concentration-time curve from zero to infinity (AUC0-∞), Assesment of drug absorption rate (Cmax/ AUC0-∞), Percent of the area measured by AUC0-t relative to the extrapolated total AUC0-∞ [(AUC0-t/ AUC0-∞) X 100].
Statistical analysis of data: Statistical analysis of the determined pharmacokinetic data was performed using statistical computerized program M-Stat software for determination of analysis of variance (ANOVA). Bioequivalence could be demonstrated for loratadine within the prescribed 90% confidence interval of 80.00% to 125.00% for AUC0-t, AUC0-inf and Cmax with respect to the parametric method on Ln-transformed data.
4. Results and Discussion
4.1 HPLC Assay
4.1.1 Analytical procedure validation
Chromatogram of loratadine: Loratadine was well separated and its retention time was 1.37 min. sharp, and symmetrical peaks were obtained with good baseline resolution and minimum tailing, thus facilitating the accurate mesurment of the peak area. The in house developed chromatographic conditions, was nearly in accordance to published literature [12,14,18] with modifications.
Linearity and quantitation: Peak area ratios of varying amounts of loratadine in human plasma (Ranging from 0.05 to 10 ng/ml) was highly linear (r2 was equal to 0.999893). The results of three replicate analysis of loratadine at three different days over one week period were obtained, where the average correlation coefficient was 0.9907, which is in accordance with the latest FDA Guidelines [16], and so it could be used for pharmacokinetic and bioavailability studies of Loratadine.
Precision and accuracy: To assess the precision and accuracy of the developed analytical method, three distinct concentrations in the range of expected concentrations where evaluated. Precision and accuracy was assessed at within-day basis, which defines those parameters during a single analytical run; and at between-day basis, which measure the between day variability, possibly involving different analysts, reagents, etc. The results of withinday accuracy of loratadine showed an average recovery percentage of 98.254%. The results of between days accuracy of loratadine showed an average recovery percentage of 100.8315%, with an average CV% of 13.027%. The results of freeze-thaw stability, short term stability, and long term stability of loratadine in human plasma showed that the average recovery of loratadine was greater than 96% providing that loratadine is stable in the studied condition.
The method used for sample preparation is in accordance with published litreature [12,18] which applies alkalinization of plasma followed by liquid- liquid extraction technique for sample preparation.
4.1.2 Bioequivalence study
Clinical observation: All the participating volunteers well tolerated the drug and the procedure adopted in the study. Every sample from the 24 volunteers during each phase was obtained at the proper. No serious adverse event, or unexpected adverse drug reaction occurred during the study. No adverse events were observed in either period.
Pharmacokinetic data and assessment of bioequivance: The assessment of bioequivalence, as a measure of efficacy, was based on pharmacokinetic pararmeters derived individually for each participant from the loratadine concentration in plasma.
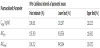
The mean Maximum plasma concentration (Cmax) was 5.8215±2.0128ng/ml and 5.3907±2.0484ng/ml, Time point of maximum plasma concentration (Tmax) 1.2778±0.8794hr and 1.4236±0.8098hr, Half-life of drug elimination during the terminal phase (t1/2e) 19.8722±3.3993hr and 20.5016±3.8831hr (Table 1), Area under plasma concentration-time curve from zero to the last quantifiable concentration estimate (AUC0-t) 48.9551±21.9226ng.hr/ ml and 46.2835±20.6650 ng.hr/ml, Mean Residence Time (MRT) 30.6343±6.2843hr and 29.6545±7.1982hr (Figure 1).


Area under plasma concentration-time curve from zero to infinity (AUC0-∞) 54.3295±22.3423ng.hr/ml and 51.3693±22.1563ng.hr/ml for test and reference products respectively (Figure 2-3). The results of loratadine pharmacokinetic parameters obtained was nearly in accordance with reported literature [19] which stated that Tmax for loratadine were found to be 1.5hr (range 0.5 to 4.5hr), Cmax 3.65± 2.82ng/ml, and reported literature [5] which stated that T1/2 ranges from 3 to 20 hours(Table 2).


Statistical analysis: The data obtained from measurments of plasma concentration was transformed prior to analysis using natural Ln tansformation. Pharmacokinetic parameters, e.g: Cmax, Tmax, AUC0-t, and AUC0-inf were analyzed using two way ANOVA procedure to rule out the possibility of a significant carryover effect. Also 90% confidence interval of 80.00% to 125.00% for AUC0-t, AUC0-inf and Cmax with respect to the parametric method on Ln-transformed data should be fulfilled. The results of 2 way ANOVA on Cmax, Tmax, AUC0-t, and AUC0-inf for loratadine showed that there was no significant difference between test and reference product (Table 3 and Table 4, Figure 4).



5. Conclusion
It can be concluded that the bioanalytical method developed for the determination of Loratadine in human plasma is valid, sensitive, specific, precise and accurate, and could be used for the determination of drug pharmacokinetic parameters. Besides, results of the bioequivalence study of loratadine oral disintegrated tablet (test product) compared to, versus the reference product are bioequivalent, since they deliver equivalent amounts of loratadine to the systemic circulation at the same rate.