


1. Introduction
The interaction between environmental and genetic factors plays a fundamental role in inducing and modulating the aging processes towards brain disorders. Among environmental factors, diet and nutritional lifestyle have a key role in brain health through the alteration of the intake or the bioavailability of metabolites and cofactors. Specific nutritional factors have particular impact on one-carbon metabolism, which is a complex biochemical pathway regulated by the presence of folate, vitamin B12 and B6 (among other metabolites) and leading to the production of S-adenosyl-L-methionine (SAM), the main biological methyl donor in transmethylation reactions, consisting in the transferring of a methyl moiety to different substrates including DNA, proteins, lipids and RNA. This metabolism involves three interrelated biochemical pathways: folate cycle, transsulfuration pathway and methionine cycle. These three metabolic sequences were first combined and correlated in 1964 by Laster’s group [1-3] giving the integrated point of view of transmethylation and transsulfuration pathways, linking sulfur amino acid metabolism to the provision of methyl groups for many biochemical processes. Normal functioning of this metabolic cycle is essential for growth and development and impairment of this metabolism; transmethylation efficiency is associated with many diseases like cardiovascular diseases [4-5], liver diseases [6-9], neural tube defects [10] and brain diseases [11-16]. One-carbon metabolism alterations, low methylation and oxidative stress are all linked to Late Onset Alzheimer’s Disease (LOAD) [17-24] Moreover, wide confirmed reports underline the role of SAM dependent reactions in age related neurodegeneration also showing the role of SAM supplementation in restoring one-carbon metabolism alterations, particularly in neurodegenerative diseases [25-29]. SAM is currently marketed as a nutritional supplement worldwide, whereas it is sold as a prescription drug in several countries of the European Union. Various evidences exist indicating that SAM has therapeutic efficacy in depression, liver diseases and osteoarthritis.
The cellular response to oxidative stress typically involves alteration of sulfur amino acids homeostasis. For these reasons, one-carbon metabolism represents the link between DNA methylation and oxidative stress [15,30]. SAM is also a precursor of glutathione (GSH), the major endogenous antioxidant. In this review we will highlight the role of SAM not just as a methyl-donor but also as a regulator of different metabolic pathways involved in the antioxidant response, underscoring the link between nutritional and genetic risk factors in brain related disorders.
2. SAM Structure and Stability
The chemical structure of SAM was first identified by Cantoni in 1951, based on the work of du Vigneaud et al. that in the late 1930s demonstrated that homocysteine (Hcy) is a product of methionine metabolism [1-2] (Figure 1). Since then, many studies have underlined the fundamental role of SAM in several important biological reactions either as a methyl donor or as an enzymatic modulator.
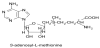
The reactivity of SAM is based on the high energy sulfonium ion that is activated for the transfer of its methyl group to nucleophilic atoms. The nucleophilic attack could be exerted by a carbon atom, as in DNA methylation; a nitrogen atom, as in glycine methylation, an oxygen atom, in catecholamine methylation or a sulfur atom, as in thiopurines methylation. SAM plays a role also in the metabolism of other substrates, like arsenic [7] and in the polyamine synthesis [2-3,6]. Referring to the stereochemical configurations of the sulfur and the alfa-carbon, respectively, SAM exists in two diastereoisomeric forms respect to its sulfonium ion, the S, S- and the R, S-SAM. Only the S, S-form is the biologically active, whereas in mammals there is a minor content of the R, S-SAM [31-32] from low to undetectable levels in many tissues, ranging from 1.5 and 3% of total SAM in mouse liver and rat brain, respectively [33-34]. This contrasts with the spontaneous S, S-SAM racemization to R,S-form occurring under physiological conditions. For these reasons, it can be hypothesized the presence in vivo of a mechanism that utilize the R, S-SAM diastereomer or transforms it to S, S-SAM.
The diastereomers separation is not workable and this leads to application of diastereomeric mixtures for SAM pharmaceutical and nutraceutical formulations: the preparations produced using the fermentation technology contain the R, S diastereoisomer in concentrations ranging from 20 to 40% of the total SAM content. This may affect its biological activity when used in in vitro and in vivo experiments, and as a food supplement or drug in clinical trials. Particular attention must be applied when handling SAM because of its high chemical and diastereoisomeric instability at specific temperatures and pH values [33,35-38]. Indeed, the cleavage to 5-methylthioadenosine (MTA) and to homoserine lactone and the hydrolysis to adenine and S-(5-deoxy-ribosyl)-l-methionine make SAM a very unstable molecule. For these reasons, SAM and SAM analogs chemical synthesis is characterized by low yields and difficulties in stereoisomer isolation. Therefore, many efforts have been made in synthesizing SAM salts formulations containing larger anions with higher stability [39-40]. Recently, in situ enzymatic synthesis was performed for stereoselective SAM analogs preparation, able to overcome these instability problems [41].
3. SAM Metabolism
An overview of the metabolic pathways involved in one-carbon metabolism is represented in Figure 2.
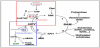
Following methyl donation to a large variety of acceptor substrates, like DNA, proteins, phospholipids, neurotransmitters and hormones, SAM is converted to S-adenosylhomocysteine (SAH), a strong competitive inhibitor of SAM-dependent methyltransferases. SAH is further hydrolyzed to Hcy and Adenosine in a reversible reaction. In fact, SAH is normally hydrolyzed to Hcy and Adenosine only if these two products are efficiently removed: Hcy accumulation causes the increase of SAH levels because of the reversibility of the reaction, since the equilibrium dynamic favors SAH synthesis. Hcy, a non proteinogenic thiol containing amino acid, is a key molecule of this pathway: it can be irreversibly converted to cysteine (Cys) and GSH by transsulfuration or remethylated to methionine by the vitamin B12-dependent enzyme Methionine Synthase (MS), that uses 5-methyltetrahydrofolate as co-substrate [1-3]. Alternatively, mainly in liver and kidney [42], betaine can donate a methyl group in a vitamin B12-independent reaction, catalyzed by betainehomocysteine methyltransferase (BHMT). The transsulfuration pathway represents the connection between sulfur amino acid metabolism and the provision of methyl groups for a large number of biochemical processes. In the transsulfuration pathway, Hcy condenses with serine to form cystathionine in a reaction catalyzed by cystathionine-β-synthase (CBS), and cystathionine is then hydrolyzed to Cys by cystathionine-γ-lyase, both vitamin B6 dependent enzymes. Cys is then used for protein synthesis, metabolized to taurine, inorganic sulphate or used for GSH synthesis [2-3,5,43]. The complete transsulfuration sequence occurs almost only in kidney, pancreas, intestine and liver, being cystathionine-γ-lyase activity tissue specific: indeed, in brain it is reported to be more than 100-fold less active than in liver [44]. In the central nervous system (CNS), Cys, the rate-limiting precursor for the synthesis of GSH production, is taken up by cells from the extracellular space, but more recent data supports the concept that transsulfuration plays a physiological role in GSH production in the brain, in astrocytes in particular: although previous studies suggested that brain tissue is deficient of the enzyme cystathionine-γ-lyase, recent works demonstrated that a functional transsulfuration pathway does exist contributing to brain GSH synthesis [43].
Regulation of metabolic flux through the competing remethylation and transsulfuration pathways occurs at several levels. When SAM is low, methionine and SAM synthesis is preserved by shifting Hcy metabolism towards remethylation, while high concentration of SAM, usually caused by methionine excess, would enhance the transsulfuration pathway. SAM and SAH are also allosteric activators of CBS, and SAM is an allosteric inhibitor of Methylenetetrahydrofolate reductase (MTHFR) [2-3,6]. SAM also inhibits MAT I and MAT II, but activates MAT III, the liver specific form of MAT (together with MAT I) [3,45].
Therefore, SAM could be viewed not only as an intermediate metabolite in methionine catabolism, but also as an intracellular control that regulates the pathway by activating or inhibiting the activity or the expression of specific enzymes, in response to injury or alterations of physiological conditions.
The alteration of one-carbon metabolism can occur either at genetic levels, with genetic defects of enzymes involved (CBS, MTHFR), or following alterations in B vitamins levels. The impairment of either remethylation or transsulfuration pathways induced by B vitamin deficiency can lead to hyperhomocysteinemia, alteration of GSH metabolism and increase of SAH levels. Being SAH a strong competitive inhibitor of SAM-dependent methyltransferases, involved in methylation of DNA (among others substrates) and gene expression regulation, SAH accumulation can induce hypomethylation, by decreasing SAM/SAH ratio (methylation potential; MP), and deregulation of gene expression [46-47]. Furthermore, the alteration of GSH metabolism derived from transsulfuration pathway impairment suggests that hypomethylation, together with the alteration of the redox homeostasis, is a mechanism through which one-carbon metabolism alteration is involved in brain diseases [46,48-49].
Due to the oxidation of the highly reactive thiol group, oxidative stress also promotes the formation of oxidized derivatives of Hcy like homocysteic acid and homocysteinesulfinic acid that generate intracellular free radicals. Moreover, Hcy can directly exert a strong oxidative damage to vascular cells and tissue, with also an indirect effect on the expression and activity of some antioxidant enzymes, like SOD and GPX [50].
Interestingly, oxidative stress is also a positive regulator of the transsulfuration pathway: oxidative stress activates CBS and inhibits MS, enhancing the conversion of Hcy to cysteine and enabling increased synthesis of GSH, to maintain cellular antioxidant defenses [1,51-52].
It seems evident that a deficiency in the transsulfuration pathway dependent on the one-carbon metabolism leads not only to a loss of cellular redox homeostasis and reduced GSH production or metabolism, but it is also strictly connected with alteration of the methylation cycle through excessive Hcy and SAH accumulation and impairment of methyl nutrients status (i.e. vitamin B12, 8 vitamin B6, folate, riboflavin, choline, methionine, cysteine, serine, glycine).
4. SAM and the Blood Brain Barrier
Studies on dose-response and bioavailability of this molecule are still scarce. Because of this lack of scientific evidences it has been questioned whether SAM crosses cell membrane and/or Blood Brain Barrier (BBB). However, there is evidence which indicates that mice treated with SAM show increased levels of both SAM and SAM/SAH ratio in plasma and brain [29] and that peripherally administered SAM is effective in the treatment of several neuropsychiatric and neurological disorders, with chronic parenteral and oral administration of SAM resulting in increased CNS levels [53]. These evidences strongly indicate that this molecule can readily reach the brain parenchyma, suggesting it penetrates the continuous layer of brain endothelial cells lining the brain microvasculature constituting the BBB. As SAM is charged at physiological pH, passive membrane permeability is unlikely to achieve the concentrations experimentally observed in the cerebrospinal fluid (CSF). The mechanism of BBB penetration by SAM has not been thoroughly investigated, however there are evidences that the blood-to-brain transfer of SAM involves transport of the complete molecule [54]. Being comprised of a methionine covalently linked to an adenosine molecule, it can be hypothesized that SAM could bypass the BBB either via the L-system aminoacid transporter or via the nucleoside transporters. In a study employing an immortalized rat brain endothelial cell line (RBE4, an established in vitro model of the BBB) the authors demonstrated that SAM is not recognized by the L-system transporter for large neutral amino acids at the brain endothelium. However, a significant interaction with the transport of adenosine indicates that SAM has affinity for the nucleoside carrier systems, suggesting that SAM may enter the CNS via the Na+-independent nucleoside carrier systems at the brain capillary endothelium [55].
5. Use of SAM as a Food Supplement or Drug
SAM level in the organism decreases with aging and restoring the original levels through exogenous supplementation is an important tool for the improvement of many vital functions.
SAM deficiency may contribute to the onset of several diseases. It is associated to the development of osteoarthritis, liver cirrhosis, depressive states and senile neurological diseases such as Alzheimer’s and Parkinson’s diseases.
Exogenous SAM is mainly administered by the oral route, although injectable formulations also exist. SAM solid oral formulations, in the form of coated tablets and capsules, are conceived to overcome the known SAM irritating activity on the gastric mucosa and to mask its unpleasant taste. For this reason, film-coated pH-dependent gastroresistant tablets are widely used in the market.
SAM is employed either as a drug or as a dietary supplement in the different countries. In the United States and Canada, SAM is sold solely as a nutritional supplement, In the USA SAM has been available under the Dietary Supplement and Health Education Act as a nutritional supplement under the marketing name SAM-e or SAMe. Also in Australia SAM is commercialized as a nutritional product.
SAM is marketed as a prescription drug under different brand names (Samyr, Adomet, Heptral, Donamet, Transmetil, Gumbal and Ademethionine), and is sold in the pharmaceutical market in Russia and CIS, India and South Korea. SAM is sold as a pharmaceutical specialty in China as well, where it is present both for oral and for intravenous administrations. In the European market, SAM is present both as a pharmaceutical specialty and as a nutraceutical product. In Italy, the dosage of 250 mg is the limit for the classification of a SAM containing product as a pharmaceutical or a nutraceutical product. Products containing more than 250 mg per tablet have to be classified as pharmaceuticals. In Mexico and Colombia, SAM can be sold in the two categories as well.
The most frequent SAM formulations available on the market are oral enteric coated tablets. Dosages of 200 and 400 mg are available on the markets. The therapeutic dose normally suggested corresponds to 800-1600 mg of SAM ion per day, often administered in several daily doses consisting of fractions of total daily dose. For preventive treatments doses of 200 or 400 mg per day are advised. In several clinical trials, the effect of SAM administered orally, has been demonstrated for doses of 200-1600 mg /day [56-57].
The use of such high doses is necessary to counteract a poor oral bioavailability. SAM is characterized by low cell permeability despite its high solubility. SAM uptake by the intestinal cells occurs by a paracellular mechanism without any involvement of membrane transporters. In vitro cellular uptake studies in Caco-2 cell cultures report how the poor oral bioavailability shown by SAM can be correlated to a limited absorption of the molecule, rather than to its rapid metabolism [58]. Efforts have been made in order to ameliorate SAM absorption by the enteric system. Some formulations, patented or under patent application, have been designed in order to improve SAM bioavailability by modifying tablet coating (US8329208, WO2011/012989, WO2010/009449, WO2015/071806).
SAM in the form of its stable p-toluensulphonate or butanedisulfonate salt has been used for more than 30 years in the treatment of depression, liver disorders, and musculoskeletal and joint disorders such as osteoarthritis and fibromyalgia.
Although the mechanism of antidepressant action of SAM is not entirely clear, it is thought that its ability to function as a methyl donor increases brain levels of serotonin, dopamine, and norepinephrine. It has been previously reported that serum and cerebrospinal fluid levels of SAM are low in depressed patients [59] and that increases in serum SAM levels correlate with improved treatment response [60]. Besides the stimulatory effect of SAM on central monoaminergic neurotransmitters, alternative mechanisms may exist in which increased or restored membrane phospholipids methylation plays a role in the antidepressant effect. SAM may increase the fluidity of cell membranes by stimulating phospholipids methylation, which has previously been linked to an increase in β-receptor and muscarinic (M1) receptor density [61]. SAM has been studied for use in various depressive disorders for many decades, with the first clinical trials dating back to as early as 1973 [62]. The majority of studies performed since then has reported that SAM is effective for treating depression, the conclusion also drawn later by a meta-analysis [63] and some other systematic reviews [56,64-65]. SAM has also been extensively studied in the context of the treatment of osteoarthritis. Experimental studies indicate that SAM increases the chondrocyte proteoglycan synthesis and proliferation rate. SAM induces the synthesis of polyamines that might stabilize the polyanionic macromolecules of proteoglycans and protect them from attack by proteolytic and glycolytic enzymes. Furthermore, in vitro studies show that SAM can antagonize the tumor necrosis factor α–induced decreases in synovial cell proliferation and fibronectin mRNA expression. These findings indicate that SAM restores basal conditions in cultured synovial cells after cytokineinduced cell damage [61]. Many trials have demonstrated that SAM reduces the pain associated with osteoarthritis and is well tolerated in this patient population.
SAM has also been used to treat various types of acute and chronic liver diseases. Although the focus of clinical trials in this area has been diffuse, a number of clinical trials have focused on the effect of SAM on cholestasis arising from a variety of causes, including pregnancy [66-68]. SAM may exert beneficial effects on the liver through a variety of mechanisms. GSH, the major anti-oxidant in the liver, plays a key role in detoxification and the limiting of oxidative damage. Studies have shown that abnormal SAM synthesis is associated with chronic liver disease, regardless of its etiology. At customary therapeutic doses, SAM has been shown to increase hepatic GSH concentrations in patients with chronic liver disease [69].
6. Involvement of SAM in Oxidative Stress and Neurodegeneration
B-vitamins and methyl nutrients are fundamental dietary factors that represent the link between nutrition and epigenetic regulation of brain disease related genes, in aging and AD [12,70]. Indeed, onecarbon metabolism alteration and consequent methylation reactions unbalance (i.e. loss of methyl groups) during aging represents one of the mechanisms by which environmental and dietary factors can promote LOAD [71-75].
Methyl nutrients participate in one-carbon metabolism and an imbalance of this metabolism could affect DNA methylation, an important mechanism for epigenetic control of gene expression, along with redox homeostasis impairment. Moreover, it is well known that methyl deficient diets induce oxidative stress that, in a sort of vicious cycle, may inhibit DNA methylation, by the oxidative stress induced DNA damage [76].
According to the theory of free radicals, physiological aging could be viewed as a gradual, inevitable process, partially generated through the accumulation of oxidative lesions. The brain is certainly the human organ most hit by aging, showing impairment yet in normal aging, and appears more prone than other organs to the occurrence of aging-related diseases. In fact, the brain is particularly vulnerable to oxidative damage and it accounts for 20% of all oxygen and 25% of all glucose consumed by the body, despite brain being only about 2% of body weight [77]. Moreover, the brain is particularly sensitive to oxidative damage for its high concentration of polyunsaturated fatty acids (PUFAs) and paucity of antioxidants as well as high concentrations of iron, and active metals which mediates the production of ROS [78-79].
Neurodegenerative disorders represent the main class of age-derived diseases and, among these, AD represents the most common disorder. There is consistent evidence in literature showing the involvement of free radical-induced oxidative damage in the etiopathogenesis of AD, since oxidative stress is one of the earliest events in AD pathogenesis [18,80-82]. Moreover, oxidative damage has been shown in the blood, CSF and brain of neurological patients with probable AD [83-85]. Post mortem studies on brain specimens collected from individuals affected by AD, evidenced extensive oxidative stress compared to healthy controls, i.e. increased levels of oxidative markers of lipids, proteins and DNA damage. The major cause of free radicals overproduction in AD seems to be related to the accumulation of misfolded protein aggregates in brain. The major component of these protein aggregates, present in the senile plaques, is amyloid beta (Aβ), a peptide of 39-42 amino acids, which derives from the sequential proteolytic processing of the amyloid precursor protein (APP) by BACE and γ-secretases. When an unbalance between Aβ production and clearance due to genetic and/or environmental factors occurs, Aβ oligomerization takes places producing different species of soluble supramolecular assemblies and some of them finally converge towards fibrillar formation. Aβ plays a central role in the pathogenesis of AD, by causing neurodegeneration and disrupting the cognitive function although the molecular pathways leading to neuronal impairment are not yet fully elucidated. It has been shown that early formed pre-fibrillar aggregates of Aβ are mainly endowed with cytotoxicity, whereas mature fibrils are much less toxic or even harmless [86-88].
In particular, soluble Aβ oligomers are associated with the generation of free radicals via direct and indirect mechanisms: in the direct one, Aβ binds to transition metals ions, acquiring an oxidase activity leading to hydrogen peroxide production; in the indirect one, neurons or microglia stimulated by Aβ oligomers produce free oxygen radicals by activation of NADPH oxidase. Notably, AD is often accompanied by a decrease of the activity and/or the levels of antioxidant enzymes such as Superoxide dismutase (SOD), Glutathione reductase, Glutathione peroxidase and Glutathione-Stransferase (GST). It is also clear that AD pathology is associated with disruptions in GSH homeostasis, the most prevalent antioxidant in brain [89].
Taken together these findings suggest a bidirectional inter-relation between Aβ and oxidative stress that may result in a vicious positive feedback pathway, and based on these evidences, a role for antioxidants in the prevention and/or treatment of AD has been hypothesized [90].
The antioxidant activity of SAM is mainly due to its role in transsulfuration as a precursor of GSH, the major cellular anti-oxidant, and as activator of CBS via transsulfuration pathway. SAM effect was largely studied in liver disease, also with attention to its antioxidant properties [9,91-92]. SAM led to inhibition of liver lipid peroxidation in a rat model of acute biliary obstruction and reduced mitochondrial oxidative stress in a rat model of liver ischemia/reperfusion [93]. SAM was also found to be able to prevent and reverse hepatotoxicity associated with several drugs such acetaminophen [94] and alcohol injury decreasing lipid peroxidation associated with tissue damage and increasing GSH levels [9]. Moreover, SAM also inhibits alcohol and lipid oxidation mainly by Fe2+ chelation and inhibition of Fe2+ autoxidation. This could represent an important mechanism by which SAM exerts cellular protective actions and reduces oxidative stress in biological systems [93].
The cell’s ability to perform methylation reactions is loosely described as “methylation potential”. Methylation status is essential for normal brain biology, and low methylation potential is associated with markers related to neurodegeneration. The importance of methylations for normal brain functions is well known. It has been recently shown that alteration of one–carbon metabolism regulates expression of two key enzymes in the amyloid pathway: β-secretase (BACE1) and presenilin (PSEN1), such that low methylation potential is associated with increased Aβ production [47,95-96]. Furthermore, activation of protein phosphatase 2A (PP2A) is SAM-dependent, and disturbed methylation status is associated with increased phosphorylation of tau, another phenomenon strictly implicated in the pathogenesis of AD [97-98]. Low methylation status is strongly associated with neurological and cognitive deficits. A chain of evidence supports the association between methylation status and neural and cognitive function. Following the discovery of an association between raised plasma total Hcy and confirmed AD, many epidemiological studies have shown that factors associated with low methylation status such as elevated total Hcy, low folate or low cobalamin levels are associated with increased risk of cognitive decline, dementia and brain atrophy. Biochemical and neurocognitive deficits related to low methylation status are potentially reversible. The efficiency of methylation therapy in restoring neurocognitive function has been extensively documented in the inborn errors of metabolism and in acquired folate and vitamin B12 deficiencies, and this beneficial effect is not confined to the childhood brain. For instance, in adult vitamin B12 deficiency, treatment rapidly improves mood and concentration ability, while reversal of neurological defects requires therapy for a longer period. Even patients with dementia may recover, particularly if their dementia is characterized by psychotic symptoms and deficits in concentration, visuospatial performance, and executive functions.
Widely confirmed reports indicate that SAM dependent reactions are compromised in age related diseases due to impaired SAM metabolism [13,26-27]. For these reasons, nutritional supplements restoring one-carbon metabolism alterations could restore physiological gene expression and oxidative buffering capacity, driving biochemical reactions to prevent or ameliorate degenerative processes. SAM diet supplementation has shown neuroprotective effects in mouse models of age-related diseases as well in clinical studies [28-29,61]. Due to the involvement of methyl nutrients metabolism in brain diseases, the role of SAM in neurodegeneration may be derived mainly from its methylating properties [27-29,47,97-98], however an important role in modulating antioxidant response in case of oxidative stress injury is recently emerged, being SAM a metabolically pleiotropic molecule participating and modulating diverse cellular processes.
In a rat model of brain ischemia/reperfusion, SAM inhibits brain lipid peroxidation, together with increased mitochondrial function [99-100]. Several experimental findings underscored the critical role of SAM in maintenance of neuronal health, suggesting a possible role of SAM as a neuroprotective dietary supplement in AD. The positive effect of SAM supplementation may be in part ascribed also to the increase in GST activity, the enzyme that plays a pivotal role in quenching oxidative species and elimination of toxic xenobiotics by forming GSH conjugates, often reduced in AD. In this work SAM supplementation restored impaired GST activity, lowering ROS levels [101].
A nutraceutical formulation consisting of six vitamins and nutraceuticals (folic acid, vitamin B12, vitamin E, Acetyl-L-Carnitine, NAC, and SAM) showed improved cognitive performance in community-dwelling adults without dementia [102], underscoring the importance of nutritional supplements in the elderly. A one-year open label pilot study examined the efficacy of vitamin/nutraceutical containing SAM formulation for early-stage AD, highlighting that to be effective, this supplementation should be started in early stage AD [103].
To study the efficacy of SAM in contrasting one-carbon metabolism alterations in neurodegeneration, Fuso and coworkers studied the pathogenic role of hyperhomocysteinemia induced by diet deficient of B vitamins (folate, B12, and B6), using transgenic TgCRND8 mice overexpressing mutated human APP gene and showing early and age-related Aβ plaque deposition [47]. The pathogenetic model was designed to reproduce, as much as possible, one-carbon metabolism alterations caused by B vitamin deficiency, inducing exacerbation of AD-like features in TgCRND8 AD mice. These effects were counteracted by SAM supplementation, through the modulation of DNA methylation and antioxidant pathways [29,95-96].
Indeed, exogenous SAM increases plasma and brain SAM levels, balances DNA methylase/demethylase activities, restores correct PSEN1 promoter methylation [95-96] and PSEN1 and BACE1 expression, reducesβ-and γ-secretase activities and Aβ production, reduces plaques to control-like values, and improves spatial memory in B vitamin-deficient TgCRND8 mice. Finally, it has also been demonstrated that the alteration of one-carbon metabolism and redox homeostasis by B vitamin deficiency and SAM supplementation was associated to tau metabolism by restoring PP2A activity and normal Tau phosphorylation [97]. Furthermore, in the same animal model SAM also prevents oxidative stress by modulating GSH metabolism and SOD activity [104]. Indeed, SAM supplementation restored normal GSH/GSSG ratio potentiating GST and SOD activity, decreasing lipid peroxidation (LPO) acting like an antioxidant. These results are in agreement with papers analyzing GSH levels and GST activity on ApoE deficient mice [101].
Interestingly, when SAM is supplemented together with an antioxidant not directly related to GSH metabolism, like Superoxidedismutase (SOD), the two molecules show synergistic effect improving both DNA methylation and the redox homeostasis and contrasting the AD-like phenotype in TgCRND8 mice [105] (manuscript in preparation). Since the cellular response to oxidative stress injury typically involves alteration in thiols content, the effect of SAM and superoxide-dismutase (SOD) supplementation on plasma thiols level was also evaluated in the same animal model, finding a significant decrease of thiols level when the B vitamin deficient diet was supplemented with SAM + SOD and SOD alone, the latter showing the greatest effect on lowering thiols [106].
Taken together, all these observations stress the role of SAM not just as a methyl-donor but also as a regulator of different metabolic pathways involved in the antioxidant response. As a nutritional supplement, SAM has been shown to prevent or alleviate hallmarks and features associated with dementia and AD, acting on DNA methylation and restoring brain oxidative status, suggesting that it could be used as a neuroprotective dietary supplement that compensates for different insults in age related neurodegenerative diseases.
Competing Interests
S. Scarpa and A. Fuso are co-inventors of a patent for the use of SAM in AD. N. Miraglia is an employee of Gnosis SpA, a pharmaceutical company producing SAM.
Author Contributions
All the authors gave substantial contributions to drafting the manuscript and revising it critically. All the authors approved the final version to be published.