


1. Introduction
Electrospun fibres have received increasing attention in the tissue engineering field, because they have a high porosity with interconnected pores, they consist of controlled fibre diameters of nano to micron size, and they exhibit flexibility. The electrospinning process utilises a high voltage source to inject electrical charge to a sample solution, such as polymer/solvent, melt and precursors of solgel glasses. The charged solution flies toward a grounded collector, resulting in the formation of electrospun fibre mats. Aligned fibre sheets and three-dimensional fibrous structurescan also be fabricated with modified electrospinning systems [1-6]. Electrospun fibre mats that are nano to submicron in diameter possess a similar structure to that of the extracellular matrix, therefore various types of electrospun fibre mats have been developed for use in tissue engineering and drug delivery systems (DDS) [7].
Fibre mats of poly (γ-glutamate) (γPGA)/silica hybrids have been developed in our previous work [8-10].γPGA is a polypeptide that has a secondary amide in the polymer backbone and is degradable by enzyme action. The carboxylic acid group, a high-reactivity functional group, is available as a polymer side chain. Jones et al. developed monoliths of the hybrids containing silica derived from (3-glycidyloxypropyl) trimethoxysilane (GPTMS) and tetraethyl orthosilicate for bone regeneration [11-13]. Hybrids consisting of the calcium salt form of γPGA (Ca-γPGA) cross-linked by silica phases derived from GPTMS are synthesized using water as a solvent[8]. The electrospun Ca-γPGA/GPTMS hybrid fibre mats are flexible and can retain their fibrous structures for several weeks in aqueous solutions. Several types of proteins, such as green fluorescent protein, chymotrypsin and thrombin, were encapsulated in the Ca-γPGA/ GPTMS hybrid fibres and successfully exhibited no denaturation and enzymic activities inside the fibres [9].
The electrospun Ca-γPGA/GPTMS hybrid fibre mats are expected to be good candidates for DDS and tissue engineering scaffolds loading growth factors, as they can encapsulate a wide range of proteins with no deleterious changes of protein structure or function because of their water-based synthesis. Denaturation of proteins can be induced by organic solvents [14]. The Ca-γPGA/GPTMS hybrid fibres have a substrate selectivity; no molecules with a high molecular weight (Mw> 1kDa) can permeate through the fibres and the permeation depends on the molecular sizes for molecules with a low Mw (Mw < 1kDa). This selectivity is expected to be of practical value in some enzyme applications because encapsulated proteins can be protected from proteases or undesirable interactions with biomolecules. Such molecule size-dependence is supposed to depend on the network structure formed by the polymer chains and the cross-linkers in the hybrids. In this study, we prepared a new type of hybrid fibre mats using (8-glycidoxyoctyl) trimethoxysilane (GOTMS) in lieu of GPTMS. GOTMS is a silane coupling agent that has the same functional group as GPTMS (epoxy group) but with a longer carbon spacer. We estimated the influences of silane coupling agents on chemical structures, degradation behaviour and molecular permeation behaviour of the hybrid fibre mats.
2. Materials and Method
2.1 Preparation of hybrid fibre mats
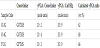
Hybrid fibre mats were prepared using the same method as described in the literature [8,9]. We mixed 0.5 g γPGA (Mw: 20,000- 50,000, Wako Pure Chemical Industries, Japan), 0.129 g of Ca(OH)2 (Kishida Chemical co., Japan) and 3 ml of ultrapure water and stirred for 0.5 h. The mole ratio of γPGA : Ca(OH)2 was 20 : 9. We added the (8-glycidoxyoctyl) trimethoxysilane (GOTMS, Shin-Etsu Silicone, Japan) to the resulting solution and stirred for 3h. The mole ratio of γPGA : GOTMS was 10 : 4 or 10 : 2.Ca-γPGA /GOTMS fibre mats were fabricated with a Nanofibre Electrospinning Unit (Kato Tech Co, Japan). The Ca-γPGA/GOTMS solution was set in a plastic syringe (10 ml, Terumo, Japan). A high tension electric field of 15-20 kV was applied to the needle (22G) and the tip of the needle was positioned 150 mm from a collector, grounded rotating drum.Fibre mats were heat-treated at 50°C for 6 h. The Ca-γPGA /GPTMS fibre mats were prepared using the same method, except GPTMS was used as a crosslinker. Sample codes of the prepared materials are shown in Table 1. Ratios of γPGA in samples (wt%) were theoretically calculated on the compositions for the preparation.
2.2 Characterisation of the fibre mats
The morphology of the prepared fibre mats was observed by fieldemission scanning electron microscopy (SEM, JSM-6301F, JEOL, Japan). We used 30 fibres in SEM images for measuring the average fibre diameter and standard deviation. Chemical structures of the prepared fibre mats were characterised by attenuated-total reflection Fourier-transformed infrared spectroscopy (ATR-FTIR, FT-IR4000, JASCO, Japan) and 29Si magic angle spinning nuclear magnetic resonance (29SiMAS-NMR, JNM-ECA600II, JEOL, Japan). The chemical shifts were measured relative to poly(dimethylsilane). To estimate the degradation behaviour of the fibre mats, 50 mg of samples were soaked in 100 ml of Tris-HCl buffer solution (pH = 7.4) and incubated at 36.5°C for 9 days.We removed 1 ml of the buffer solution from the sample solution and replenished with a fresh solution at each time point. The removed sample solution was analysed for Ca and Si concentrations by inductively coupled plasma atomic emission spectrometry (ICP-AES, ICPS-7000, Shimadzu) (n = 3).To calculate the ratio of released amount of Ca and Si relative to their total amounts in the samples, the fibre mats were completely dissolved with NaOH aq. (20 mM) and the Ca and Si concentrations of the resulting solution were measured with ICP-AES.
2.3 Preparation and characterization of fibre mats encapsulating fluorescent molecules
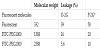
Fibre mats encapsulating fluorescent molecules were prepared usinga similar method to that described above, except molecule solutions were added to the hybrid solution before electrospinning. Three different types of fluorescent molecules (Table 2) were added to carbonate-bicarbonate buffer solution (20 mM, pH = 9.0), resulting in formation of a molecule solution (3 mM of molecule concentration). We added 500μl of the prepared molecule solution to the hybrid solution 0.5 h before electrospinning and then stirred for 0.5 h (total mixing time: 3 h). Electrospinning for the hybrid solution containing the molecule solution was performed using the method described in the previous section. Characterisation with SEM and ATR-FTIR was carried out for the prepared fibre mats encapsulating molecules. For estimation of molecule leakage from the fibre mats, 1.5 mg of the prepared samples was immersed in 3 ml of Tris-HCl buffer solution (20 mM, pH = 7.5) for 1 h.After removing the fibre mats from the solution, the absorbance (A492) was monitored to determine the ratio of leakage for each encapsulated molecule (n = 3).
*Data from reference 9.
3. Results and Discussion
Figure 1 shows SEM images of the prepared hybrid fibre mats. All of the prepared fibre mats consisted of submicron-sized fibres. The average diameters of O-2G,O-4G and P-2G were 186±18, 273±33 and 222±51 nm, respectively. The diameter of O-2G was smaller than that of P-2G. Since chemical reactivity of functional groups in silane coupling agents are influenced by the variable length of the spacers, the ratio of crosslinking formed in the hybrid systems and/or viscosity of the solution used for electrospinning would be different between O-series and P-series, causing the observed differences in fibre diameters of the resulting fibre mats [15,7].

Figure 2 shows ATR-FTIR spectra of the fibre mats and raw chemicals used for preparing them. Peaks in all of the spectra of the fibre mats were similar to each other. Bands at 1700–1740 cm−1 were attributed to C=O vibration of carboxyl groups [16]. Peaks at around 1740 cm−1 were attributed to crosslinking between epoxy groups in GOTMS or GPTMS and carboxyl groups in Ca-γPGA [17]. Si-O-Si bonds, which were formed due to condensation between silanol groups in GOTMS or GPTMS, which was observed in the fibre mats[18]. Peak shifts at around 1100 cm−1 were found between O-series and P-series; the peaks for O-2G appeared at a higher wave number than P-2G. These peak shifts are likely to be due to changes in siloxane structures formed between O-2G and P-2G. This is supported by the 29Si MAS-NMR spectra (Figure 3) and the results of the degradation tests (Figure 4). In the 29Si MAS-NMR spectra, the Tn sites indicate a silicon atom bonding to one carbon atom (Si-C) and to n bridging oxygen bonds (CSi(OSi)n(OH)3-n)[19]. Both O-2G and P-2G spectra indicate the presence of T1(−50 cm−1), T2(−60 cm−1) and T3(−70 cm−1) species. Since no T0 peak was observed, most of the silanol groups in the samples were found to bond with each other (condensation). The largest peak area in the O-2G spectrum was that of T3, whereas the peak areas of T2 and T3 were similar to each other in the P-2G spectrum. This indicates that siloxane phases in O-2G consist of more condensed siloxane than P-2G.



Figure 4 shows the percentage Si and Ca release of elements contained in the fibre mats. The O-series exhibited a burst of Si and Ca release at12 h after soaking. Although the percentage values of Si release were different between O-2G and O-4G, those of Ca release were almost the same. Releases of both elements from O-series stopped after the burst and the percentage Si and Ca release values remained equal. In the case of P-2G, the Si release behaviour was clearly different to that of the O-series; the percentage release values gradually increased with the immersion time and continued to increase until the end of the immersion test (~9 days). The Ca release exhibited a burst at12 h after soaking and then its percentage Si and Ca release values gradually increased with time. The burst of Si release from the O-series may be attributed to the GOTMS that did not contribute to the crosslinking in the hybrid system. Most of them are expected to have formed siloxane, sinceno T0 peak was observed in the 29Si MAS/NMR for O-2G. The amount of GOTMS that formed free siloxane phases in the hybrids varied with the GOTMS amounts used, since the percentage values of Si release from O-4G were almost twice those from O-2G. However, most of the GPTMS contributed to the crosslinking in the hybrid system because the Si release of P-2G was gradual.The total amounts of Si release from P-2G, however, were more than four times those from O-2G. This indicates that the crosslinking was more stable with GOTMS than GPTMS. More condensed siloxane phases formed in the O-series might contribute to the lower degradation behaviour. Although the Si release behaviour was different between O-2G and O-4G, the Ca release was almost the same between the two samples. Therefore, Ca2+ ions must coordinate to carboxyl groups in the polymer chains instead of the siloxane phase.
The results of the molecule leakage test are shown in Table 2. Low Mw molecules were able to permeate the O-2G fibres like P-2G and their leakage level was dependent on their Mw. However, the percentage leakage decreased when changing the cross-linker from GPTMS to GOTMS.We further confirmed there were almost no differences in fibre diameters and chemical structures of the molecules encapsulated in the samples between SEM observations and ATRFTIR spectra (Figure 5). Encapsulating the molecules was found not to influence the fibre morphology and the chemical structure of the hybrids.

Although further investigations will be needed to clarify the effects of cross-linkers on the molecule leakage, we expect that the reduction of the molecule leakage was due to the varied swelling behaviours of the hybrid system in the buffer solution. Cross-linked γPGA is one of the super absorbent polymers [20]. These super-absorbent polymers are polymer-based hydrogels that can absorb water at >95 % of the total weight or volume. Some hydrogels exhibit controlled release of encapsulated proteins, drugs etc. [21]. Their release depends upon swelling and shrinking of hydrogels. Therefore, both O-series and P-series hybrid fibres are expected to absorb water (swell) after soaking in the buffer solution and then release encapsulated molecules. Since GOTMS contains a longer carbon chain, which reduces hydrophilicity of the hybrid fibres, the swelling of the O-2G fibres was suppressed and the molecule permeation in the fibres was inhibited. In addition, the network mesh size, which is formed by cross-linked polymer chains in hydrogels, is an important parameter for controlling the swelling of hydrogels and the release of encapsulated proteins and drugs [21].
The network mesh size in the hybrid fibres is supposed to change as a result of using different cross-linkers, because the O- and P-series hybrid fibres vary in both the length of carbon chains in their molecules and in the rate of condensation of siloxane phases formed. This is likely to have induced the varied molecule leakage observed in the present study. The varied swelling behaviour was also expected to have influenced their degradation rate. Thus, the degradation rate and the leakage of molecules encapsulated in the fibres were controlled by the use of GOTMS instead of GPTMS.
4. Conclusions
We compared two different types of the γPGA/silica hybrid fibre mats prepared using GOTMS or GPTMS as cross-linkers. The degradation behaviour and the release rate of the fluorescent molecules were smaller for the γPGA/GOTMS hybrids than the γPGA/GPTMS hybrids. The chemical durability and the molecule release behaviours of the hybrids were controllable using proper cross-linkers, which will be useful for developing ideal materials for tissue engineering and DDS.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
We would like to thank ORTHOREBIRTH Co. Ltd. (Japan) for their helpful discussion in part in developing the materials. We thank Shin-Etsu Silicone (Japan) for kindly providing GOTMS.