


1. Virology and Entry into a Host Cell
To understand the origins of the COVID-19 pandemic we must first remember the severe acute respiratory syndrome coronavirus (SARSCoV) which emerged in 2002. The two virus strains, SARS-CoV and SARS-CoV-2 are similar; phylogentic analysis has demonstrated that SARS-CoV-2 has approximately 76% nucleotide identity with SARSCoV [1]. Work by Hoffman et al. has shown that SARS-CoV-2 enters an identical spectrum of cell lines as SARS-CoV and its mechanism of entrance is similar. This suggests similarities in choice of entry receptors for both viruses. Both viruses depend on their spike (S) proteins for entry into a host cell. The S1 unit of the S protein attaches and binds to the host cell’s receptor. Entry into the cell then requires priming of the S protein by the host cell’s cellular proteases which leads to a fusion of the viral and host cell’s membranes leading to endocytic entry. The host cell receptor is angiotensin- converting enzyme 2 (ACE2) and the serine protease employed is TMPRSS2 [1]. This entry causes cellular internalization of ACE2 and downregulation of these receptors: the importance of which will be described later. Normally, ACE2 is only scarcely present in the circulation in a soluble form; however, ACE2 is widely expressed and its receptors have been found in the arterial and venous endothelial cells and arterial smooth muscle cells of nearly every organ, including the lung, heart, kidney, and brain; and has been found in abundance in the oral and nasal mucosa, lung alveolar epithelial cells, enterocytes of the small intestines, cardiac myocytes, renal podocytes and cells of the proximal convoluted tubule [2-5]. This may, in part, help explain the array of systemic symptoms seen in COVID-19.
2. Proposed Pathogenesis of SARS-CoV-2 in the Lung
We propose that the virus likely first enters the host through the oral or nasal mucosa and then spreads either down to the gastrointestinal system or into the respiratory system before causing systemic infection in some patients. There is a wide spectrum of the severity of respiratory manifestations in patients infected with SARS-CoV-2; some patients may have a dry cough for a few days, while others suffer from acute respiratory distress syndrome (ARDS) requiring mechanical ventilation with high positive end expiratory pressures (PEEPs). The progression to ARDS may be spurred by the following mechanisms, illustrated in Figure 1. ACE2 is found in abundance on the alveolar epithelial cells, particularly in pneumocyte type II cells [5]. Though the exact mechanisms are unclear, based on the current literature, we hypothesize that cellular internalization and subsequent spread of the virus in the respiratory system leads to the following processes: 1) an increase in the ratio of levels of angiotensin converting enzyme 1 (ACE1) and angiotensin II compared to levels of ACE2 and angiotensin 1-7; 2) a significant inflammatory response mediated by neutrophils, macrophages and CD8+ T cells leading to alveolar edema; 3) thrombus formation; 4) potential destruction of the pneumocyte type II cells [2,5-12].

Decreased ACE2 levels play a significant role in the progression to ARDS. ACE2 inactivates angiotensin II while generating angiotensin 1-7, thus it downregulates the rennin-angiotensin system (RAAS) [2,6]. The subsequent, enhanced and unopposed, vasoconstriction may, in part, explain the atypical nature of the ARDS that develops in COVID-19. What we have witnessed in our institutions is similar to what has been observed by Gattinoni et al. in Italy; namely, that the lungs which develop ARDS retain a relatively high level of compliance. Despite reasonable lung compliance, severe hypoxemia persists even when using recruitment strategies such as high PEEP and prone positioning [13].
Dysregulated lung perfusion and hypoxic vasoconstriction via loss of ACE2 and the consequent increase of angiotensin II, the potent vasoconstrictor, to levels of angiotensin 1-7 can help explain some of the hypoxemia. Increased pulmonary inflammation and coagulation have been reported as effects of unopposed angiotensin II. In a study involving 12 patients, it was found that angiotensin II levels in the plasma samples from SARS-CoV-2 infected patients was markedly elevated and linearly associated to viral load and lung injury [14]. Further aggravating this change in the ratio of Angiotnesin 1-7 to Angiotensin II, is that angiotensin II upregulates disintegrin and metalloproteinase 17 (ADAM17) which cleaves membrane-anchored ACE2 [5]. This causes a loss of the catalytic activity of ACE2 on membranes. Elevated levels of circulating soluble ACE2 are a sign of increased activity of the RAAS system and are associated with a worse prognosis [5]. Additionally, loss of ACE2 can cause neutrophil accumulation which eventually contributes to ARDS and increased vascular permeability and thusly pulmonary edema [2]. This has clinical significance as it may inform future treatment strategies. For instance, studies in mice infected with avian influenza H5N1, showed that administration of recombinant human ACE2 decreased virusinduced lung injury [15]. Of additional interest, is that angiotensin II further interferes with adaptive immunity by activating macrophages, and other immune cells, causing increases of IL-6, TNF-alpha and other inflammatory cytokines [5].
SARS-CoV-2 infection results in reduced levels of peripheral CD4+ Tcells and CD8+ T cells though they remain hyperactivated [14,16]. CD8+ T cells are highly cytotoxic. Dysregulation of T cells leads to alveolar barrier destruction. Studies on SARS-CoV showed that the virus had an affinity for lymphocytes and numbers of both CD4+ Tcells and CD8+ T cells were reduced [14]. In a cohort of 191 patients, Zhou et al. found that COVID-19 non-survivors had a significantly lower baseline lymphocyte count than survivors [17]. A relative disruption in the number of CD4+ T cells, often times the gate keepers of the immune response, leads to dysregulation of the entire immune system [7]. Elevated levels of biomarkers interleukin-6 (IL- 6), tumor necrosis factor-alpha (TNF-α), among others, are a sign of severe disease, and are associated with increased mortality [7]. CD8+ T cells damage the alveolar epithelial cells by secreting cytotoxic granules including granzymes that enter the cytoplasm of the target cell and trigger the caspase cascade, which eventually leads to apoptosis [18]. Apoptosis is also induced by cell-surface interaction between the TC and the infected cell by the Fas ligand on the infected cell binding to the Fas molecule that is expressed on the T cell [18]. Furthermore, damage to the alveolar type II cell in the lung has multiple consequences including inactivation of surfactant production, which worsens barrier disruption and alveolar edema and leads to accumulation of protein-rich fluid within the interstitium and alveoli [5,18]. Additionally, loss of surfactant has a detrimental effect on the elasticity of the lung tissue.
The immune dysregulation that occurs in some patients infected with SARS-CoV-2 has been termed, “Cytokine storm.” This term, since the beginning of the pandemic, has been oft used but also poorly understood. Cytokine release syndrome (CRS), another name for this disorder, occurs in various conditions, including bacterial sepsis, in patients treated with chimeric antigen receptor (CAR) T cell therapy and hemophagocytic syndrome [19,20]. CRS causes increased systemic inflammation, increased vascular permeability which contributes to the formation of pleural effusions and edema; CRS also leads to intravascular depletion via third space fluid loss and hypotension [20]. IL-6 is perhaps the most important cytokine in CRS. It has been found that in patients infected with SARS-CoV-2 elevated levels of IL-6 correlate with development of ARDS, adverse clinical outcomes and death [19-21].
Increased coagulation activity, marked by increased d-dimer concentrations, in some patients with COVID-19, further disrupts oxygenation. High levels of d-dimer have a reported association with increased mortality [17,22]. Inflammation and endothelial damage further increases procoagulant factors causing further endovascular damage and potentially eventual thrombosis and ischemia [8-12]. Laboratory test findings of 3 patients infected with SARS-CoV-2 at Tongji Hospital included leukocytosis, elevated prothrombin (PT) and partial prothrombin time (PTT), elevated fibrinogen and d-dimer levels, the presence of anticardiolipin IgA antibodies, antibeta2- glycoprotein I IgA and IgG antibodies [12]. Most of these antibodies are part of antiphospholipid syndrome and can lead to thrombotic events in rheumatological diseases. They can also rise in response to various viral infections such as hepatitis C virus, human immunodeficiency virus, cytomegalovirus, varicella zoster, Epstein- Barr virus, adenovirus, and parvovirus B. In many instances, the presence of these antibodies was associated with thrombosis [12,23,24]. This is a potential mechanism of thrombosis in patients suffering from COVID-19 as well. IL-6, among other interleukins, is known to cause platelet hyperactivity, clumping and aggregation [25]. Furthermore, tumor necrosis factor (TNF) mediated expression of tissue factor promotes platelet aggregation and multifocal thrombi formation and interalveolar coagulation [25]. This occurs because TNF activates vascular monocytes and vascular endothelial cells to express tissue factor on their cell surfaces. This activates the extrinsic pathway of the coagulation system to induce microthrombus formation. Another pathway by which TNF contributes to microthrombus formation is by decreasing the endothelial expression of thrombomodulin and glycosaminoglycans that regulate the coagulation system [23,25]. All these dysregulations can lead to disseminated intravascular coagulation (DIC). TNF and other cytokines activate neutrophils to release various inflammatory mediators such as neutrophil extracellular trap, histones, leukotriene B4, MMP matrix metalloproteinase, MPO myeloperoxidase, reactive oxygen species and others which are capable of further damaging the endothelial cells [23].
Another potential mechanism of hypoxia was proposed by Liu et al. [26]. After performing a molecular analysis, it was discovered that COVID-19 possesses ORF8, ORF10, orf1ab, ORF3a and surface glycoproteins that interact with heme component of the hemoglobin. Each heme group contains iron ions that are attached to porphyrin. These iron ions participate in oxygen and carbon dioxide exchange in the body. ORF8 proteins have a capability to bind to the porphyrin in the heme. ORF10, orf1ab and ORF3a proteins can dissociate iron from the heme molecule of beta 1 hemoglobin chain (see figure 2). This way hemoglobin loses its ability to effectively deliver oxygen [26] and therefore some of the circulating hemoglobin becomes nonfunctional, contributing to further hypoxia. This is analogous to carbon monoxide poisoning. The dysfunctional hemoglobin causes the red blood cells to lose rheology and become deformed. This is potentially another contributing factor to hypoxia and thrombosis in the microcirculation. It is postulated that hydroxychloroquine inhibits the de-ironization of iron from heme [20], thereby preserving the oxygen carrying capacity of hemoglobin. Although the initial injury may occur in the lungs, the systemic inflammation and hypoxia have severe effects on other organs like the heart and kidneys. The evidence regarding this mechanism is rather limited at this time; both experimental and clinical trials are needed.

3. Proposed Pathogenesis of SARS-CoV-2 and the Heart
The exact mechanisms of cardiac injury in COVID-19 are not well established but the following outlines some of the likely causes. The cardiac effects are partially due to the reduction of ACE2, inflammatory responses, hypoxia, and disruptions in the coagulation pathway (see Figure 3). ACE2 is widely expressed in cardiomyocytes, cardiac fibroblasts and coronary endothelial cells [2]. The increase of angiotensin II relative to angiotensin 1-7 due to COVID-19 has many deleterious effects in the heart. Angiotensin 1-7 exhibits antiproliferative, antiapoptotic, and mild vasodilating abilities and also has protective effects including those protective against heart failure, thrombosis, myocardial hypertrophy, fibrosis, arrhythmia, atherogenesis and also attenuates vascular dysfunction related to metabolic syndrome [2]. Other viral diseases like influenza and respiratory syncytial virus (RSV) have been associated with cardiac complications and poor outcomes [10,27]. There are several autopsy reports that showed acute myocardial infarction in patients infected with SARS-CoV-2. Zhou et al. reported that in a cohort of 191 patients infected with SARS-CoV-2, increased high-sensitivity cardiac troponin I was found in more than half of patients who died [17].

Shi et al. found that in a study of 416 patients, those with cardiac injury had a higher mortality than those who did not, 51.2% vs 4.5% [28]. It was found to be an independent predictor of mortality with a hazard ratio of 4.26 [28]. The potential causes are many-fold. Systemic inflammatory response can lead to increased inflammatory activity within coronary atherosclerotic plaques. This, in conjunction to endothelial dysfunction, increases the risk of plaque rupture [28]. Conversely, in studies in which patients, infected with SARSCoV- 2, were found to have favorable troponin levels, they also more commonly had favorable outcomes [28,29]. The inflammatory response likely plays a large role in the myocarditis that develops in some patients infected with SARS-CoV-2. Autopsy reports have shown inflammatory infiltrates including an abundance of macrophages and some CD4+ T cells [7]. Though, as of this writing, there have not been reports of SARS-CoV-2 within myocardial tissue; however, viral genome was found in 35% of patients, in one study, who died from SARS-CoV [30].
Due to the difficulty of obtaining regular echocardiography’s in patients with COVID-19, it is not possible to say with confidence whether there is a greater propensity of heart failure with preserved ejection fraction (HFpEF) or reduced ejection fraction (HFrEF). However, heart-failure symptoms are common in patients suffering from COVID-19. Zhou et al. described heart failure in 23% of their cohort, and in 52% of their non-survivors. In addition to the potential direct effects of COVID-19, the inflammatory response, the prothrombotic state that arises due to the disease, one must also take into the consideration the treatments currently being offered to patients infected with SARS-CoV-2, many of which increase the QTc interval. This includes but is not limited to the anti-malarial medications chloroquine and hydroxychloroquine, the antiviral agents, and the antibiotics such as azithromycin.
4. Proposed Pathogenesis of SARS-CoV-2 and the Kidney
Inflammation, vasoconstriction and the propensity for thrombosis following SARS-CoV-2 infection are systemic in nature and affect the kidneys in many ways. As more studies are published regarding COVID-19, the incidence of acute kidney injury (AKI) in patients infected with SARS-CoV-2 will be clearer. Co-expression of the ACE- 2 and TMPRSS genes has been found to be no less in the kidneys than that of the lungs and this suggests that kidney cells may be an important target for SARS-CoV-2 [3]. Current literature suggests that SARS-CoV-2 displays a tropism towards the kidneys [31]. As of this writing, work by Yang et al., has shown that in a study of 52 critically ill adult patients, 23% suffered from AKI [32]. Zhou et al. found that AKI was found in 15% of their cohort overall, and in 50% of their non-survivors. There is relatively high co-expression of ACE2 and TMPRSS in podocytes and in the proximal straight tubules. Podocyte and proximal tubule destruction can explain why proteinuria and hematuria was seen in many patients with COVID-19 on presentation, 43.9% and 26.7%, respectively, in a prospective cohort study of 701 patients by Cheng et al [33]. Previous studies on ACE-2 and renal cells in mice models has shown that inhibition of ACE2 leads to increased albuminuria and glomerular matrix expansion, increased mesangial matrix deposition, glomerular basement membrane thickening and glomerulosclerosis [34]. On the other hand, amplification of ACE2 has been shown to reduce the effect of nephropathy in mouse models [4]. Additionally, the inflammatory response described previously may play a profound effect on the kidneys (see Figure 4).

Moreover, if renal tubular epithelium is injured, it may worsen cytokine overproduction [20]. The mechanism of this is not well understood; however, it has been observed that injury to tubular epithelium cells promotes the upregulation of IL-6 in human and animal studies [20]. Also of interest, is that podocytes are one of few cells to express IL-6 receptors on their surface thus they can directly respond to IL-6, and these receptors are upregulated during inflammatory processes [35]. We propose that this upregulation of the immune response in turn can cause further lung injury which may in turn lead to more profound hypoxic renal medullary injury. Worsening cardiac function can also explain some of the AKI that occurs, as cardiomyopathy and myocarditis may ultimately lead to renal vein congestion and renal arterial hypoperfusion. These mechanisms, along with fluid imbalances, high airway pressures may lead to a renal compartment syndrome [20]. The disruption of the coagulation pathway and thrombus formation as described earlier likely also plays a role in the development of AKI in many patients with COVID-19. Additionally, as with all critically ill patients with a dysregulated immune response, the risk of superimposed bacterial infections, especially in patients with foley catheters, is high and warrants monitoring and consideration.
5. Current Understanding of Therapeutic Targets and Treatment Options
Figure 5 summarizes the proposed four pathophysiologic mechanisms of injury that culminates into organ damage following SARS-CoV-2 infection and what is currently in consideration as an approach to therapy. It is not clear why 80% of the population will clear the virus at onset but others progress to downstream effects provoking strong inflammatory response and vasoconstriction that then increases the risk for thrombotic events and organ injury. Nonetheless, it appears that the inflammatory and vasoconstrictive responses are tightly linked and occur simultaneously. The inflammatory and vasoconstrictive responses, along with a hypercoagulable state induced by procoagulants produced as a result of the infection is a perfect setup for microcirculatory thrombosis and ultimately end organ damage. In the lungs this leads to acute respiratory distress syndrome (ARDS), with ventilation perfusion mismatches and explains why the ARDS in these patients does not respond as expected to high pressure mechanical ventilation support systems. To address COVID-19, there is opportunity to treat with agents that directly inhibit SARS-CoV-2 replication, such as antivirals (remdesivir)[36], macrolide antibiotics such as azithromycin and the anti-malarial hydroxychloroquine37 with caution [38], but randomized clinical trials to promote the widespread use of the agents as best practice are not yet available. The use of convalescent plasma [39] shows promise and is in clinical trials. It is important to note the pyrexia and diarrhea seen in these patients induces significant fluid deficits that must be replaced; however, care must be taken to not overhydrate these patients because of the risk of worsening ARDS. The ultimate goal in SARS-CoV-2 infection prevention, is vaccine development [40] for which studies are underway. To address the inflammatory response, steroids [41] and the selective IL-6 blocker (Tocilizumab) [42] are in use but response is variable. The full spectrum of how the cytokine storm may be approached from lessons learned in influenza H1N1 is detailed by Liu et al. [43] and how immunosuppressive drugs affect COVID-19 response to therapy, including the use of JAK kinase inhibitors, is also thoroughly reviewed by Russel et al [44]. The use of non-specific anti-inflammatory or cell depleting agents is likely to attenuate host response, exacerbate viremia and should be discouraged. More recently the concept of using mesenchymal stem cells [45] for immunomodulatory and regenerative effects in COVID-19 infected patients is gaining consensus for clinical trials. To address the vasoconstriction, research into fusion inhibitor to block the virus from entering cells and administration of exogenous ACE2 to ameliorate the effects of angiotensin II seems prudent [15,45]. Preliminary observations of angiotensin II converting enzyme inhibitors (ACEI) angiotensin type 1 receptor blockers (ARB) in hospitalized patients have shown reduction in mortality [46] and therefore opens up another facet for clinical trials to be conducted. To address the thrombosis, all patients who progress to mechanical support and elevated d-dimers should be anticoagulated [47]. In our earlier encounter with these patients, this was not a consideration until more recently evidence in favor of anticoagulation has changed our management of these patients. The role of exchange transfusion to replace dysfunctional hemoglobin caused by de-ironization of heme in COVID-19 infection is yet to be determined and whether hydroxychloroquine helps stabilize iron in hemoglobin [48] thereby contributing to improvement in oxygenation is unclear. Finally, better understanding of the pathogenesis along with early interventions are critical in avoiding end organ damage. End organ damage requires support. In ARDS, lung protective ventilation (avoiding high positive end expiratory pressures) with high flow oxygen delivery, as well as physical therapy methods including being in the prone position [49] are the main stay of therapy, while in AKI, dialysis may be required. The use of cardiac support devices is limited in managing SARSCoV- 2 infected patients, nonetheless as we gain better control of each check point and the ability to confirm conversion to negative tests, opportunities will avail for studies in cardiac support that may pave a way for lung transplantation or combined heart and lung transplantation. The concept of using mesenchymal stem cells [45] for immunomodulatory and regenerative effects in SARS-CoV-2 infected patients for lung repair following ARDS is gaining consensus for clinical trials.
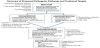
It is important to emphasize that none of the therapeutics for SARSCoV- 2 infection described here have been tested in randomized clinical trials and are at best investigational or used compassionately. It is also important to note that our attempt is to outline potential mechanisms of organ injury in SARS-CoV-2 and where treatments may be targeted and not an in-depth review of available treatments for COVID-19.
6. Conclusion
Despite the short experience of COVID-19 pandemic and acknowledging that much of the pathogenesis is yet to be uncovered, significant strides have been made in our current understanding of the pathogenesis of injury that has paved the way for us to illustrate the potential therapeutic targets. Each therapeutic target has an existing treatment options, but none has been approved for widespread use. Therapies to slow viral replication and certainly vaccine development for prevention are critical, nonetheless, it appears that a multi-faceted approach addressing some or all the proposed therapeutic targets is necessary to prevent or slow end organ injury. At this point, it is prudent to start treating a patient with COVID-19 based on or current understanding of pathogenesis, until future research unveils more insights into the pathogenesis and randomized clinical trials point us in a more structured direction for best practice.
Competing Interests
The authors declare that they have no competing interests.