


1. Introduction
Niosomes are unilamellar or multi-lamellar vesicles, wherein an aqueous solution is enclosed by a bilayer structure made up of nonionic surfactant with or without cholesterol [1]. Niosomes can entrap both hydrophilic and lipophilic drugs in aqueous core and vesicular membrane, respectively. The bilayers of niosomes have both inner and outer surfaces to be hydrophilic with sandwiched lipophilic area in between. Thus, a large number of drugs and other materials can be delivered using niosomes [2]. Although niosomes were tried for various routes such as intramuscular [3], intravenous [4], peroral and transdermal [5], it is used in the market for transdermal delivery of various drugs. Studies showed that an enhanced delivery of drugs was observed when niosomes were utilized. Niosomes have been reported to enhance the residence time of drugs in the stratum corneum and epidermis, while reducing the systemic absorption of the drug and improve penetration of the trapped substances across the skin. In addition, these systems have been reported to decrease side effects and to give a considerable drug release [6]. They are thought to improve the horny layer properties both by reducing transepidermal water loss and by increasing smoothness via replenishing lost skin lipids [7]. In addition, as drug delivery vesicles, niosomes have been shown to enhance absorption of some drugs across cell membranes [8], to localize in targeted organs [9] and tissues and to elude the reticuloendothelial system.
In the recent years, niosomes received great attention as an alternative potential drug delivery system to conventional liposomes. Moreover, compared to phospholipid vesicles, niosomes offer higher chemical and physical stability with lower cost and greater availability of surfactant classes [7,10,11].
The properties of the vesicles can be changed by varying the composition of the vesicles, size, lamellarity, tapped volume, surface change and concentration [12].
The bilayers of the niosomes protect the enclosed active pharmaceutical ingredient from the deterogenous factors present both inside and outside the body. So, niosomes can be used for the delivery of labile and sensitive drugs. Niosomes are osmotically active and stable and also increase the stability of the entrapped drug. The surfactants used and also the prepared niosomes are biodegradable, biocompatible and non-immunogenic. Handling and storage of surfactants does not require any special conditions [12].
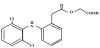
Aceclofenac belongs to non steroidal anti- inflammatory drugs (NSAIDs) [13]. It is an orally administered phenyl acetic acid derivatives, with a molecular weight 354.18 g/mole and a chemical designation of 2-[2-[2-[(2, 6-dichlorophenyl) amino] phenyl] acetyl] oxy] acetic acid (Figure 1).
It is an effective analgesic and anti-inflammatory agent with a good tolerability profile. It works by blocking the action of cyclooxygenase. Cyclooxygenase is involved in the production of various chemicals in the body, some of which are known as prostaglandins. Prostaglandins are produced in response to injury or certain diseases and would otherwise go on to cause pain, swelling and inflammation [13]. Aceclofenac has also shown stimulatory effect on glycosaminoglycan synthesis in human osteoarthritisc cartilage [14] and chrondro protective effect mediated by the suppression of metalloprothenases production and proteoglycan release in rabbit articular chrondrocyte and human rheumatoid synovial cell [15]. Aceclofenac has proved as effective as diclofenac, piroxicam in patients with osteoarthritis, rheumatoid arthritis and in patients with ankylosing spondylitis. It provides effective analgesia in other indications such as dental or gynaecological pain, lower back pain and ear, nose and throat indications [16].
Prolonged use of the drug is associated with gastro intestinal disturbances such as GI discomfort, nausea, diarrhea and peptic ulceration [17].
All the drugs in this group reduce inflammation caused by the body's own immune system and are effective pain killers. But it has several drawbacks such as narrow therapeutic index, short biological halflife [13]. These factors necessitated the development of niosomal gel formulation for aceclofenac. This dosage form would reduce the dosing frequency, and enhance patient compliance.
The present study was aimed to develop aceclofenac niosomal gel followed by evaluating different parameters such as, encapsulation efficiency, shape, particle size distribution, in-vitro drug release and skin permeation. In-vivo performance of gel containing drug loaded niosomes compared with gel containing free drug was evaluated by assessing anti-inflammatory activity using carrageenen-induced rat paw edema test.
2. Materials and Methods
2.1 Materials
Aceclofenac was generously gifted by SmithKline Beecham Cairo, Egypt. Hydroxyl propyl methylcellulose (HPMC), Pluronic F-127, Carbpol 934, carrageenan and semi-permeable cellulose membrane (12000-14000 MWCO) were purchased from Sigma Chemicals, St. Louis, MO, USA. Sodium alginate and sodium carboxymethyl cellulose were obtained from BDH Ltd (Poole, UK.). Chloroform, methanol, potassium dihydrogen phosphate, disodium hydrogen phosphate and sodium hydroxide were obtained from United Company for Chem. and Med. Prep., Egypt. Diethyl ether, Span 20, 60 and 80, propylene glycol were purchased from Adwic, El-Naser chemical co., Egypt. All chemicals were of analytical grade and used as received.
2.2 Methods
2.2.1 Preparation of niosomes
Aceclofenac niosomes were prepared using the reverse-phase evaporation technique [18,19]. Accurately weighed quantities of surfactant (span 20, 60, 80) and cholesterol equivalent to 50 mg were mixed in 250 ml long-necked quick fit round bottom flask and dissolved in 10 ml chloroform. The organic solvent was slowly evaporated under reduced pressure, using a rotary evaporator, at 40°C to produce a thin lipid film. The lipid film was redissolved in 10 ml diethyl ether, and the aceclofenac dissolved in 5 ml acetone mixed with 5 ml distilled water. The mixture was sonicated for one minute, swirled by hand, and resonicated for another minute. The organic solvents were evaporated on the rotary evaporator under reduced pressure for two hours. The niosomes were allowed to equilibrate at room temperature. The niosomal dispersion was kept in the refrigerator to mature overnight (4°C). The noisomal formulations of desired qualities are listed in Table 1.

2.2.2 Characterization of aceclofenac niosomes
2.2.2.1 Determination of drug encapsulation efficiency
For the separation of non-entrapped aceclofenac, the nisomal dispersion was subjected to centrifugation on a refrigerated centrifuge (Hettich, Germany) at 14000rpm at 4°C twice, each of 30min duration with 10min gap to separate free drug from entrapped drug. After centrifugation, the supernatant was siphoned off carefully to separate the non-encapsulated aceclofenac leaving behind the niosomes with the entrapped drug in the sediment.
The niosomes were separated from the supernatant and 0.1ml of the supernatant was added to10ml methanol in a 10ml volumetric flask, and the amount of drug was determined spectrophotometrically at ?273nm. The pellet obtained after centrifugation was lysed in methanol and sonicated for 10 min. The concentration of aceclofenac was determined in methanol spectrophotometrically at ?273 nm.
Percentage drug encapsulation (EE %) was calculated as follows:
EE % = [(T-C)/T] ×100
Where T is the total amount of drug that is calculated both in supernatant and sediment, and C is the amount of drug found only in the supernatant [19,20]. In all cases aceclofenac-free niosome formulation was used as a blank. The average of three different measurements ± SD is reported.
Several factors affecting the encapsulation efficiency of aceclofenac loaded niosomes were investigated to find out the optimal conditions for preparation of niosomes. These factors are the effect of added drug content, the effect of cholesterol weight ratio and type of surfactant (Table 1).
2.2.2.2 Determination of niosome size and size distribution
The vesicle size and size distribution were determined using a laser diffraction technique on a Mastersizer X Ver. 2.15 (Malvern instruments Ltd. Malvern, UK). The measurements were performed at 25°C, using a 45mm focus lens and a beam length 2.4mm. The particle size analysis was carried out on freshly prepared niosomal dispersions of different weight ratios (Table 2).
2.2.2.3 Photomicroscopic analysis
Samples of aceclofenac-loaded niosomal formulation (N-2) were examined microscopically at magnification of 1000x with a binocular microscope equipped with a camera (Motic, Japan). A drop of niosomal suspension placed on microscopic slide was examined and photographed.The morphology and structure of the same niosomal preparation, (N-2) were observed using transmission electron microscopy. The niosomal dispersion was diluted 10-folds using deionized water. A drop of diluted niosome dispersion was applied to a carbon-coated 300 mesh copper grid and left for 1min to allow some of the niosomes to adhere to the carbon substrate. The remaining dispersion was removed by absorbing the drop with the corner of a piece of filter paper. After twice rinsing the grid (deionized water for 3–5s) a drop of 2% aqueous solution of uranyl acetate was applied for 1sec. The remaining solution was removed by absorbing the liquid with the tip of a piece of filter paper and the sample was air dried. The sample was observed with a transmission electron microscope (JEOL 100 CX, Japan) operated at 80KV [21].
2.2.2.4 Fourier-transform infrared spectroscopy (FT-IR):
Fourier-transform infrared spectroscopy (FT-IR) measurements were performed using Hitachi 295 spectrophotometer using KBr disc method. The samples were scanned over the range of 4000 to 400cm-1. Infrared spectroscopic analysis was done for cholesterol, drug, surfactant, and physical mixture of them at same amount.
The excipients such as surfactant, cholesterol, pure drug and physical mixture of excipients and the drug were mixed separately with infrared (IR) grade KBr in the ratio of 1:100 and corresponding discs were prepared by applying 15000lb of pressure in a hydraulic press. The discs were scanned in an inert atmosphere over a wave number range of 4000-400cm-1 in Hitachi 295 spectrophotometer (Hitachi, Tokyo, Japan). FT-IR analysis was also performed for niosomal dispersion as a thin liquid film applied on KBr disc.
2.2.2.5 Differential Scanning Calorimetry (DSC) Measurements
Differential scanning calorimetric studies were carried out using differential scanning calorimeter (TA-60, Shimadzu, Japan) for the samples (Aceclofenac, surfactant, cholesterol and aceclofenacloaded niosomes). For this purpose 3mg of each sample were sealed thermatically in standard aluminum pans. Each sample was scanned between 30 - 200°C using nitrogen as the purge gas. For calibrating enthalpy, indium was sealed in aluminum pan with sealed empty pan as a reference. Thermograms were obtained at a scanning rate of 10°C/ min.
2.2.3 Preparation of aceclofenac gel formulations
Selected aceclofenac-loaded niosomes (equivalent to 0.5% w/w drug) were incorporated into different gel bases. Gels containing 0.5% w/w aceclofenac dissolved in polyethylene glycol 600 (20% w/w) were also prepared for comparison. The polymers used were carbopol 934 (1% w/w), pluronic F-127 (20%w/w) and HPMC (3%w/w), sodium carboxy methyl cellulose (3%w/w), and sodium alginate (7%w/w).
The required quantity of carbopol 934 was weighed and dispersed in a small amount of distilled water to prepare an aqueous dispersion. The aqueous dispersion was allowed to hydrate for 4-5hours. Drug (0.5%w/w) was added and properly dispersed. The pH was adjusted to 6 by addition of 1% (w/v) triethanolamine solution. The final weight of the gel was adjusted to 10g with distilled water.
Pluronic gels were prepared by the cold method as described by Schmolka [22]; the calculated amount of the polymer was dispersed in cold distilled water, and the solution was left in a refrigerator overnight until the mixture became clear solution. The drug was then added and thoroughly mixed into the pluroinc solution. The final weight of the gel was adjusted to 10g with distilled water.
HPMC, sodium alginate and sodium carboxy methyl cellulose gels were prepared by dispersing the required quantity of polymer in small quantity of distilled water to prepare an aqueous dispersion. The aqueous dispersion was allowed to hydrate for 4-5hours. Drug (0.5% w/w) was added and properly dispersed then the final weight of the gel was adjusted to 10g with distilled water.
Niosomal gels were prepared using the same procedure. For this purpose calculated amount of niosomal suspension containing 50mg drug was centrifuged and the pellets obtained were incorporated into the gel by vortex. Vortexing was continued until a homogenous niosomal gel was obtained; the gel was then sonicated to become bubble-free.
2.2.4 Viscosity measurement
The viscosity of different formulations was determined at room temperature using Brookfield DV+II model LV viscometer. The measurements were made using spindle S-96 at 1.5 rpm.
2.2.5 In-vitro drug release
A piece of standard cellophane membrane was stretched over one end of an open-ended glass tube which was made water tight by the use of a rubber band. The tube was immersed in a vertical position inside a 150 ml beaker containing 50 ml of phosphate buffer (pH 7.4) and was maintained in a thermostatically controlled shaker (shaked at 50 stroke /min in a water bath maintained at 37±1°C to reflect the usual skin temperature).
One gram of niosomal gel formulation, equivalent to 5mg of aceclofenac was placed inside the release tube over the cellophane membrane. At predetermined time intervals for up to 6h, 5ml aliquots of the release medium were withdrawn for analysis and were replaced with equal volume of release medium at the same temperature to maintain constant volume. The absorbance of the collected samples was measured spectrophotometrically at ?max 273nm. The results are the mean values of three release experiments ±SD.
2.2.6 In-vitro skin permeation
In-vitro permeation through rat hairless skin was performed using the same procedure described above for the in-vitro drug release except that the abdominal skin of Wister rats (weighing 200-250 g) was used as permeation membrane. The epidermal surface was facing upward and allowed to be in contact with phosphate buffer pH 7.4. The stratum corneum was facing the donor side of the cell. At predetermined time intervals (up to 6 h), samples of 3 ml were withdrawn from the receptor medium and replaced with an equal volume of fresh medium. Samples were analyzed spectrophotometrically at ?max = 273 nm. Each experiment was repeated 4 times and the average ± SD were calculated. The cumulative amounts of drug permeated (mg/cm2) were calculated and were plotted versus time (h). Aceclofenac steady state flux (Jss) was calculated as the slope of linear regression line at the steady state phase for each experimental run. Apparent permeability coefficient (Papp) was calculated according to the following equation:
Papp = JSS/C0
Where, C0 is the initial drug concentration.
2.2.7 In-vivo anti-inflammatory efficacy of aceclofenac gels
The anti-inflammatory effect of the selected gel formulations were evaluated using the paw edema test in rats. Experiments were carried out according to the animal ethics guidelines of Assiut University, Egypt. Male albino rats weighing 200 ± 20g were divided into 3 groups (6 per group). Rats were fasted with free access to water for 24h prior to the test. The animals of group 1 (control) received 1g of placebo gel (HPMC gel without drug), groups 2 and 3 received 1g of HPMC gel containing free drug, and HPMC gel base containing aceclofenacloaded niosomes (N-2), respectively. For the induction of edema, a volume of 50µl of 3.0% (w/v) carrageenan suspension in 0.9% saline solution was injected into the sub plantar region of the right hind paw of the rat [23]. The thickness of the right paw for each rat was measured by Vernier Caliper (SMEC, China) before carrageenan injection and immediately after carrageenan injection (0 time) and then every hour for up to 6 hours. The percent swelling of the paw was calculated using the following equation [24]:
% Swelling = [(V - Vi)/Vi] x 100
Where V is the paw thickness at each time interval, and Vi the initial paw thickness before carrageenan injection.
The average paw swelling in the drug treated rats was compared with that of control rats (which received placebo) and the percent inhibition of edema was determined using the following relation [24]:
% Inhibition = [1 - (% swellingtreated / % swellingcontrol)] x 100
where swellingtreated is the mean value observed in the treated group, and swellingcontrol is the mean value observed in the control group.
2.2.8 Statistical analysis
Statistical analysis of the obtained results was carried out by the student t-test at 0.05 level of significance.
3. Result and Discussion
3.1 Characterization of aceclofenac niosomes
3.1.1 Factors affecting the encapsulation efficiency of aceclofenac into niosomes
The factors influencing the encapsulation efficiency of aceclofenac into niosomes were investigated to find out the conditions for maximum entrapment of the drug into niosomes.
3.1.1.1 Effect of added drug content
Different drug amounts were added to evaluate their effect on percent encapsulation. Surfactant: cholesterol weight ratio of (0.5:0.5) was chosen to evaluate the effect of the addition of 5, 10 and 15 mg aceclofenac on percent encapsulation upon preparing niosomes using reverse-phase evaporation method.
Table 1 showed that, for niosomes prepared from span 20, 60 and 80, increasing the drug loading from 5 to 15mg resulted in significantly higher entrapment efficiency (p < 0.05).For niosomes prepared from span 20 there was no significant increase in entrapment efficiency when the drug loading increased from 10 to 15 mg. This could be attributed to the saturation of the bilayers of span 20 niosomes at drug loading of 10mg.
So, generally, increasing the drug content from 5mg to 15mg led to higher entrapment efficiencies for almost all formulae. This is attributed to the higher concentration of drug in the organic phase during the reverse evaporation process, which would lead to larger amount of aceclofenac intercalated in the bilayer, that agrees with the result reported for amphotericin B inclusion into liposomes prepared by reverse-phase evaporation method [25].
3.1.1.2 Effect of cholesterol weight ratio
Cholesterol is a common component of niosomes providing rigidity to the membrane, controlling permeability and improving plasma stability in a dose dependent manner [26]. The effect of cholesterol on drug encapsulation efficiency was evaluated by preparing the niosomes at three surfactant to cholesterol weight ratios, namely 0.5:0.5, 0.5:1 and 0.5:1.5. The entrapment efficiency for each niosomal dispersion was estimated.
The effect of cholesterol on aceclofenac entrapment was different according to the non-ionic surfactant used. For span 20 and span 60 niosomes, nonsignificant increase in entrapment efficiency was showed when cholesterol weight ratio increased from 1 to 2 weight ratios (p > 0.05). This may be due to the following two factors:
- With increasing cholesterol, the bilayer hydrophobicity and stability increased [27] and decreased the permeability [28] of the bilayer preventing more entrapment of aceclofenac within the bilayer, as the niosomes form.
- In contrast, higher amounts of cholesterol may compete with the drug for packing space within the bilayer, hence excluding the drug as the amphiphiles assemble into vesicles. Another explanation to the above mentioned situation is that, increasing cholesterol beyond a certain concentration can disrupt the regular linear structure of the vesicular membrane [29].
On the other hand, span 80 niosomes, a significant increase in entrapment efficiency occurred when cholesterol weight ratio increased from 1 to 2 weight ratios (p < 0.05). This is because span 80 has the lowest transition temperature due to unsaturated double bond in the oleate side chain above the phase transition temperature. So, cholesterol made the membrane more ordered and abolishes gel to liquid phase transition and it was able to prevent leakage of the drug from niosomes and more stability occurred [30,31].
3.1.1.3 Effect of surfactant type
Trails have been made to prepare and get good niosomes containing surfactants having HLB values between 14 to 17 such as Tween 20, 60 and 80, Brij 35 and Myrj 52 but failed to obtain the required product. In all the niosomes prepared with spans, as the concentration of surfactant increased, drug entrapment efficiency increased. Data in Table 1 reveals that the entrapment efficiencies for niosomes prepared using span 60 were superior to those prepared using span 20 and 80. This can be explained by many facts: (a) the hydration temperature used to make niosomes should usually be above the gel to liquid phase transition temperature of the system that results in niosomes that are less leaky and have high entrapment efficiency. Span 60 has highest phase transition temperature (50°C) as compared to span 80 and 20 and hence high entrapment efficiency, (b) The length of alkyl chain of surfactant has a prominent effect on permeability of prepared niosomes as length of surfactant increases entrapment efficiency also increases . Hence, long chain surfactant results in high entrapment. Thus, span 60 has a longer saturated alkyl chain (C16) compared to span 20 (C10), so it produces niosomes with higher entrapment efficiency. (c) The longer alkyl chain influences the HLB value of the surfactant mixture which in turn directly influences the drug entrapment efficiency. The lower the HLB of the surfactant the higher will be the drug entrapment efficiency and stability as in the case of niosomes prepared using span 60 [32]. Also, span 60 having critical packing parameter (CPP) ranging from 0.5 to 1 entrap drug molecules without any cholesterol [13].
Formulations of span 20 have higher entrapment efficiency in comparison to span 80 formulations. This could be attributed to the structure of surfactants; span 20 has the saturated alkyl chain while comparison to span 80 formulations. This could be attributed to the structure of surfactants; span 20 has the saturated alkyl chain while span 80 has an unsaturated alkyl chain. The introduction of double bonds made the chains bend. This means the adjacent molecules cannot be tight when they form the membranes of niosomes. This cause the membrane to be more permeable, which possibly explains the lowest entrapment efficiency of span 80 formulations. It suggests that the alkyl chain is a critical factor of permeability and saturated chain produces higher entrapment [33].
3.1.2 Determination of niosome size and size distribution
The particle size of aceclofenac niosomes was studied using laser diffraction technique. The values were obtained by averaging of three measurements at 25°C.
3.1.2.1 Effect of surfactant type on particle size
Table 2 shows that the niosomes prepared using span 60 is larger in size than those prepared using span 20 and span 80. Span 60 has a longer saturated alkyl chain compared to span 20 as mentioned previously, and it was reported that surfactants with longer alkyl chains generally give larger vesicles. This would account for the higher entrapment efficiencies with niosomes prepared from span 60, also the mean size of the niosome increased with progressive increase in the HLB value because surface free energy decreases on increasing hydrophobicity of surfactant. Span 20 has shortest alkyl chain cause smallest particle size [32].
3.1.2.2 Effect of surfactant concentration on particle size
Average vesicular diameter values for span 20, span 60 and span 80 formulations containing the same amount of drug and cholesterol were presented in Table 2. The data clearly indicate an inverse relationship as the amount of surfactant increased there was a decrease in vesicular diameter [34].
Increasing hydrophobicity of the surfactant monomers led to smaller vesicles, a result which is expected since surface free energy decreases with increasing hydrophobicity [35].
3.1.2.3 Effect of cholestrol concentration on particle size
Table 2 revealed that, in case of span 20 and span 60 niosomes by increasing cholesterol concentration led to increase in particle size. This because cholesterol increases the width of bilayer and consequently increases the vesicle size [34]. Also, this occurred in case of span 80 niosomes due to presence of double bond [36].
3.1.3 Photomicroscopic analysis
The aceclofenac niosomes prepared in this study visually appeared as white suspension. The photomicrograph of aceclofenac niosomes (N-2) composed of span 20: cholesterol (0.5:0.5) weight ratio is shown in figure 2. Niosomes prepared by reverse-phase evaporation method reveal the presence of homogeneous population of unilamellar vesicles with one hydrophopic bilayer and oligolamellar vesicles consisting of a few concentric bilayers. The aceclofenac entrapped vesicles are spherical in nature, having large internal aqueous core and exist in disperse and aggregate collections.

The transmission electron micrograph of aceclofenac niosome is shown in figure 3. It can be seen that the particles are spherical in shape and have a smooth surface, a fairly uniform size and having a definite internal aqueous space [20].

3.1.4 Fourier-transform infrared spectroscopy (FT-IR)
Drug-excipient interaction was studied before developing the formulation by using FT-IR spectroscopy, which is one of the most important analyses to investigate the stability of formulation, and molecular interactions between the drug and the used excipients.
FT-IR spectrum of aceclofenac (Figure 4a) showed intense band of functional groups, 3319.3 cm-1 (N-H stretching), 3271cm-1 (O-H stretching), 2970 cm-1 (C-H stretching) and 1716.58cm-1 (C=O stretching ). The absorption band at 743.4cm-1 (C-H bending), 1253.88 cm-1 and 1346.48 cm-1 (C-N stretching) and 1450.06 cm-1 (O-H bending).
The IR spectrum of pure cholesterol (Fig ure 4b) showed characteristics peaks at 3377.74cm-1, (aliphatic O-H stretch), 2935.29 cm-1, (C-H stretch) and 1715. 89 cm-1, (carbonylic C=O stretch of ester).
The IR spectrum of span 60 (Fig.4c) showed characteristics peaks at 3383.43 cm-1, (aliphatic O-H stretch), 2925.31 cm-1 (C-H stretch) and 1742.93 cm-1 (carbonylic C=O stretch of ester).
The IR spectrum of the physical mixture of aceclofenac with cholesterol and span 60 (Figure 4d) showed their additive spectra.
In case of medicated niosomes (N-8), IR spectrum showed minor shifting of some peaks compared with individual excipients and the drug such carbonylic C=O stretch of ester (1716.58 to 1731.40) and O-H bending (1450.06 to 1455.26) (Figure 4e).

These shifts may be due to the formation of hydrogen bonds, Van der Walls attractive forces or dipole moment which are weak forces seen in the polar functional groups of drug and excipients. The frequency of absorption due to the carbonyl group depends mainly on the force constant which in turn depends upon inductive effect, conjugative effect, field effect, stearic effects. The shifts seen due to the above mentioned interaction may however support the formation of favorable vesicle shape, structure with good stability and sustained drug release [37].
3.1.5 Differential Scanning Calorimetry (DSC) measurements
This kind of study may be of interest for the preparation of niosomes with high drug entrapment capacity and with suitable drug-release characteristics. The interaction was studied by DSC, which represents a non-invasive analytical technique.
DSC analysis was carried out for aceclofenac, span 60, cholesterol and aceclofenac –loaded niosomes (N-8). The DSC thermogram of aceclofenac showed sharp endothermic peak at 156.10°C which corresponds to the melting of the drug. Span 60 and cholesterol showed endotherms at 59.10°C and 128.40°C, respectively. DSC thermograms of aceclofenac-loaded niosomal dispersion, interestingly exhibited endotherms at 90.46°C and 103°C. The cholesterol endotherm exhibited a shift from 128.40°C to 103°C, while endotherm at 90.46°C indicated the increase in phase transition temperature of niosomes upon loading with aceclofenac. Absence of the melting endotherm of aceclofenac suggested that drug changed from crystalline to amorphous state (Figure 5). These results suggest significant interaction of drug with the bilayer structure and can account for the enhanced entrapment of aceclofenac into niosomal formulations and sustained drug release [38,39].

3.2 In-vitro drug release
Release of aceclofenac from different niosomal gels was investigated to find out the suitable conditions for optimum formulation of acelofenac niosomal gel. Niosomal formulae were chosen on the basis of highest entrapment efficiency and lowest particle size. The niosomal formulae were (N-2), (N-8) and (N-13) (Table 1 and 2), were incorporated in different gel bases, such as carbopol 934, sodium alginate, sodium carboxymethyl cellulose (NaCMC), hydroxypropylmethyl cellulose (HPMC ) and pluronic F-127. Figures 6-10 show the release of aceclofenac from niosomal gel formulations.





It was found that, in all release studies, carbopol 934 (1%w/w) and pluronic F-127 (20%w/w) gels showed lower release than sodium alginate (7%w/w), sodium CMC (3%w/w) and HPMC (3%w/w) gels. The release pattern of aceclofenac could be ranked as: HPMC > sodium alginate > sodium CMC > carbopol 934 > pluronic F-127 gels. Generally, niosomal gels showed lower release of aceclofenac than non-niosomal aceclofenac gels.
The release of aceclofenac from niosomal gels is a combination of the release of drug from niosomes and the diffusion through the polymer network channel structures of the gel. The entrapped aceclofenac molecules could leak out gradually from the vesicles into the surrounding gel. This finding is in agreement with other previous studies [40,41] which reported that the cholesterol present in the monolayer would limit 1, 2-dipalmitoyl-sn-glycero- 3-phosphoethanolamine (DPPE) mobility and favor an upright orientation of the DPPE hydrocarbon chains thus enhancing Van der Waals interactions. Consequently, it would contribute to a spontaneous mixing between the two membrane lipids and to an increased stability of the monolayer.
Also, inclusion of cholesterol within the niosomal structure could markedly reduce the flux of aceclofenac during the release phase, which is in accordance with the membrane stabilizing activity of this lipid [42].
Moreover, this rank of drug release is mostly reflective of the viscosity of gel formulations (Table 3). Viscosity of the gel formulation is a very important factor that affects the release of drug since it may reduce diffusion rate of drug from the vehicles. Hereby, a reverse relationship was observed between the viscosity of liposomal gels and the percent of aceclofenac released. This is in agreement with many previous studies [43,44].

(Mean ± SD, n = 3)
For HPMC (3%w/w) niosomal gel, the cumulative percent drug release at 6 hours for the span 20 is 84.79%. The cumulative percent drug release at 6 hours for the span 60 is 82.24%. The cumulative % drug release at 6 hours for span 80 is 52.88 %. Significant changes in release were observed upon changing the type of surfactant used in the bilayer of aceclofenac niosomes (p < 0.05) (Figure 6).
3.3 In-vitro skin permeation study
The effect of surfactant type used in the preparation of aceclofenac loaded niosomes can be tested in this study as aceclofenac was delivered from HPMC (3%w/w) niosomal gel. The enhanced delivery through the stratum corneum of noisome encapsulated drugs has been observed [45] and it therefore remains to elucidate the mechanism of this delivery, especially as the stratum corneum is considered to be a particularly impermeable barrier [46].
Figure 11 shows the cumulative amount of aceclofenac permeated (mg/cm2) across rat skin employing niosomal and non-niosomal HPMC gel From this figure, it could be concluded that, first, the niosomal gels showed higher skin permeation of aceclofenac through the excised hairless rat skin which was significant than the nonniosomal gels (p < 0.05). Second, a correlation existed between the type of surfactant and the amount of aceclofenac permeated through skin membrane.

Permeation parameters are presented in Table 4. In-vitro permeation parameters revealed that the highest flux of aceclofenac was 0.075 mg/cm2/h in case of niosomal gel containing span 20, followed by niosomal gel containing span 60 (Table 4). Lower flux values were obtained by using non-niosomal gel which clearly vouches for the permeation enhancing effect of vesiculation on the drug. The flux values were 3.7 - 2.2 times higher from the niosomal gels contain span 20 and span 60 respectively, compared to non niosomal gels. The highest mean permeability coefficient of aceclofenac was obtained from HPMC niosomal gel contain span 20 (1.43 ± 0.10 cm/h).

(Mean ± SD, n = 3)
Several mechanisms could explain the ability of niosomes to modulate drug transfer across skin [47,48], including: 1-Adsorption and fusion of niosomes onto the surface of skin would facilitate drug permeation, 2-The vesicles act as penetration enhancers to reduce the barrier properties of the stratum corneum, 3-The lipid bilayers of niosomes act as a rate-limiting membrane barrier for drugs.
One of the possible mechanisms for niosomal enhancement of the permeability of drugs is structure modification of the stratum corneum. It has been reported that the intercellular lipid barrier in the stratum corneum would be dramatically looser and more permeable following treatment with liposomes and niosomes [48,49]. Both phospholipids and nonionic surfactants in the proniosomes can act as penetration enhancers, which are useful for increasing the permeation of many drugs. Fusion of noisome vesicles to the surface of skin, demonstrated in a previous report [49], results in higher flux of the drug due to direct transfer of drug from vesicles to the skin.
3.4 In-vivo anti-inflammatory efficacy
The anti-inflammatory effect of aceclofenac from the HPMC (3%w/w) niosomal gels contain N-2 niosomes, HPMC contain free drug and HPMC gel were assessed, to study the effect of formulation on the pharmacologic effect.
Based on the in-vitro release and skin permeation studies, HPMC (3%w/w) based gels were selected for further anti-inflammatory efficacy evaluation. Niosomal gels containing span 20 (N-2) were selected for further anti-inflammatory efficacy evaluation. Three groups of male albino rats were used, the first group was considered as a control and received HPMC (3%w/w)) gel, the second group received one gram of free non-niosomal aceclofenac gel and, the third received one gram of aceclofenac niosomal gel (N-2). The gels were applied to the right hind paw of the rats. The thickness of right paw of each rat was measured before carrageenan injection, immediately after carrageenan injection (0 time) and then after (0.5, 1, 2, 3, 4, 5, and 6 hours).
Statistical analysis between two groups using (t-test paired two sample for means) was done and showed that the anti-inflammatory effect of niosomal gel is significantly higher (p < 0.05) and the effect sustained for longer time than free aceclofenac gels. This coupled with superior skin penetration and deposition potential of niosomal gel formulation which definitely indicated depot forming properties of niosomal gel. Hence, perhaps better skin permeation potential and depot characteristic of niosomal gel formulation resulted in better inhibition of paw edema.
Table 5 shows the time course of percent swelling and percent edema inhibition by aceclofenac niosomal and non-niosomal gels. Induction of acute inflammation in control rats resulted in a prominent increase in paw thickness beginning 0.5h after carrageenan injection.

Mean ± SD, n = 6
When aceclofenac gels were applied locally on the edematous paw, after the first 2h of the observation, HPMC niosomal gel showed 59.31% edema inhibition while that in case of non niosomal gel was 52.43%. After 6 h, the inhibitory effect was 85.89 % and 37.16 % for the same gel formulations, respectively.
At all times, niosomal gel showed higher edema inhibition than nonniosomal gel. The inhibition continued to increase till the end of the experiment in case of niosomal gel. While in case of non-niosomal gel, the % edema inhibition started to decline after 3h. As compared with the control group treated with placebo, both formulations were highly effective (p < 0.001), with a significant difference between them (p < 0.05). These results show that aceclofenac niosomal gels are more effective in reducing the edema than aceclofenac non-niosomal gels, also the anti-inflammatory effect of niosomal gels was sustained for longer time (up till 6h).
4. Conclusion
In conclusion, the results of the present study demonstrated that HPMC niosomal gel formulation containing span 20: cholesterol (0.5:0.5) possesses great potential for prolonging release, enhancing skin permeation and improving and sustaining the local antiinflammatory effect of aceclofenac. So, this formulation seems to represent an attractive strategy for sustained delivery of aceclofenac.