


1. Introduction
In 2012 14,1 million new cancer cases have been diagnosed, cancer caused the death of 8,2 million patients and 32,6 million people are living with cancer (within 5 years of diagnosis) world-wide [1].
Gynaecological tumours are among the most common cause of cancer death and currently causing more than 100,000 deaths per year [2]. The highest tumour-associated mortality of gynaecological malignancies is related to ovarian cancer [2]. Therefore ovarian cancer is an important public health problem and there has been no appreciable improvement in survival for women with ovarian cancer over the past 40 years. The survival of ovarian cancer is poor and more than 70% of cases are diagnosed at late stage.
In ovarian cancer treatment platinum-based chemotherapy plays a pivotal role as first line chemotherapy option in combination with taxane [3]. Therefore platinum-resistance is the most crucial problem for treating ovarian cancer and there is a definite clinical need to develop new treatment strategies to overcome platinum resistance.
Increasing evidence points towards AKT over-expression and alteration of the PI3K/AKT/mTOR cascade as a mechanistic reason for this resistance.
This review provides a short overview of the PI3K/AKT/mTOR signalling network, the interplay with the Ras/Raf/MEK/ERK signal cascade and discusses the rationale for targeting the PI3K/ AKT/mTOR pathway in cancer with a special focus on tumour immmunological aspects. A better understanding of the molecular mechanism causing cancer therapy-resistance could most probably result in new therapeutic options able to revert chemotherapy resistance and enhance sensitivity to platinum-based chemotherapy drugs in ovarian tumours.
2. Phosphatidylinositol-3-Kinase (PI3K)/AKT Signal Transduction Pathway
One of the most frequently altered signalling pathways involved in cancer as well as in resistance development especially in ovarian cancer is the PI3K/AKT/mTOR pathway (Figure 1).
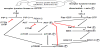
This pathway is a central signal transduction pathway, which transmits signals from multiple cell surface receptors to transcription factors in the nucleus [29-31]. Furthermore there exist several direct interactions between PI3K/AKT- and Ras/Raf/MEK/ERK-signal transduction pathways (Figure 1) [32].
The Ras/Raf/MEK/ERK signal cascade can be activated via binding of several extracellular components like growth-factors or adhesion molecules to receptor tyrosine kinases [28]. The receptor tyrosine kinase activation results in dimerisation and auto-phosphorylation [33]. The growth-receptor-bound-protein 2 (Grb2) binds to the receptor dimer and causes transformation of Ras-GTPase from an inactive state with bound GDP into active state with bound GTP. The activated Ras kinase binds and activates than the serine-threoninekinase Raf. Phosphorylated and by this activated Raf in turn activates MEK via phosphorylation and the phosphorylated MEK has the capacity for ERK and MAPK activation [28,34-36]. ERK1 and ERK2 phosphorylate a huge range of nuclear and cytoplasmatic substrates resulting in initiation of different cellular responses involved in proliferation, differentiation, cell cycle progression and cell death [33].
3. Crosstalk Between the PI3K/AKT- and Ras/Raf/MEK/ERK-Signal Transduction Pathways
Different studies have shown that an over-expression of AKT results in down-regulation of ERK [12,37]. Furthermore crosstalk between these pathways was observed after treatment of cells with pharmaceutical inhibitors of PI3K which decreased ERK activity [38-41]. This interaction suggests cooperation between the two pathways in order to determine functional outcomes.
The Ras superfamily of small G proteins is one of the connecting points between the PI3K/AKT/mTOR- and Ras/Raf/MEK/ERKsignal transduction cascade. It was shown that Ras is involved in activation of PI3K [32,42,43].
Furthermore it was demonstrated that also the PI3K/AKT-pathway controls the Ras/Raf/MEK/ERK-signal transduction cascade; phosphorylated AKT decreased the Raf activity [43-45].
The protein p70S6K was identified as another connecting point between the PI3K/AKT/mTOR- and Ras/Raf/MEK/ERK-signal transduction cascade [46,47]. The serine-threonine-kinase p70S6K is downstream of PIP3 and PDK1 in PI3K/AKT/mTOR-pathway and a direct target of mTOR and also a direct target of ERK1/2 [47,48]. Of special interest is that activation of p70S6K is involved in feed-back inhibition of the PI3K/AKT/mTOR-signal transduction cascade [49].
4. Alteration of the PI3K/AKT/mTOR-signal Transduction Pathway in Tumours
Recent studies indicate that numerous components of the PI3K/ AKT/mTOR-pathway are targeted by amplification, mutation and translocation more frequently than any other pathway in cancer patients with resultant activation of this pathway [22].
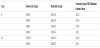
Both genetic and biochemical data suggest that activation of the PI3K/AKT/mTOR survival pathway contributes to ovarian cancer development and tumour genesis [17]. Such activation is caused by different mechanisms and one mechanism is somatic alterations in PI3KCA that have been found in a substantial fraction of ovarian cancers [50]. PIK3CA amplifications are present in 40% of ovarian cancers [21]. Furthermore, activation of PI3K/AKT/mTOR signal transduction pathway is caused by mutations in the gene coding for PIK3CA. Frequencies of PIK3CA mutations in subtypes of ovarian cancer are shown in table 1 and in table 2 the frequencies of specific PIK3CA mutations in ovarian cancer are summarized.

Another alteration that results in increased activity of the PI3K/ AKT/mTOR pathway is PTEN loss-of-function. PTEN loss is observed in about 7% of all ovarian cancer cases and it seems to be more common in type I ovarian tumours [51-56].
For AKT a point-mutation in the PH-domain has been detected in ovarian cancer [57]. This point-mutation results in conformational change of the PH-domain so that AKT can be activated without the presence of PI3K [57].
Also the deregulation, mutation or over-expression of cell surface receptors can result in an increased activity of the PI3K/AKT/mTOR signalling pathway in ovarian cancer [58]. Furthermore Ras mutations are found in 20% of low-grade ovarian cancers [59]. Because of the fact that Ras has been shown to activate both the Ras/Raf/MEK/ ERK and the PI3K/AKT/mTOR pathway, mutation of Ras should theoretically activate both pathways simultaneously. But up to now it is not evaluated in detail if Ras mutations result also in an increased activity of the PI3K/AKT/mTOR-signalling pathway. Nevertheless one study exists that demonstrates that some Ras mutations result in deregulated PI3K and downstream AKT activation [60]. Beside Ras mutations also the over-expression of different other proteins e.g. Rab25 [61], Twist2 [62] or MyD88 [63] seems to result in activation of AKT. The fact that AKT can be activated by a huge number of different proteins underlines the importance of AKT under physiological and pathophysiological conditions. Nevertheless, in human specimens of ovarian cancer AKT was found to be activated in 68% [64].
5. Effects of Altered PI3K/AKT/mTOR-signal Transduction Pathway in Tumours
As mentioned before, AKT is an important junction known to regulate various cellular pathways that promote cell survival, cell proliferation, angiogenesis and invasion. Furthermore, the epithelialmesenchymal- transformation, an important step for tumour metastasis, has been shown to be related to AKT activation [65]. Deregulation of components of the PI3K/AKT-cascade not only contributes to ovarian cancer development and tumourigenesis but also to chemotherapeutic drug and radiation resistance as it was recently shown [4,20,66-81]. The sensitivity of cells to radiation and chemotherapeutic drug-induced apoptosis is determined by the balance between cellular survival and apoptosis [4,14]. Due to the well-known anti-apoptotic role of AKT, an AKT over-expression in cancer cells might most probably mediate resistance to radiation and chemotherapy.
Beside the PI3K/AKT/mTOR signalling cascade other general mechanisms of resistance exist. In this review other possibilities of platinum-resistance will be only shortly mentioned. Some publications describe that platinum-resistance can be also caused by differential expression of microRNAs [82-84] as well as by detoxification of bioactive platinum-complexes by sulphur-containing peptides or proteins, cellular compartimentation and increased DNA repair [66]. Furthermore diminished drug accumulation caused by reduced uptake or increased efflux of platinum compounds via heavy metal transporter can result in platinum therapy failure [66].
Recently it was demonstrated that microRNAs involved in platinumresistance are directly involved in regulation of PTEN, AKT or other downstream molecules of the PI3K/AKT pathway [85-92].
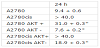
The fact that members of the PI3K/AKT/mTOR pathway are regulated by microRNAs involved in platinum-resistance emphasises the importance of the PI3K/AKT/mTOR signalling cascade as therapeutic target. Therefore inhibition of PI3K/AKT/mTOR signalling in ovarian carcinomas seems to be a promising target to enhance the efficacy of anticancer agents such as cisplatinum and to pathway are regulated by microRNAs involved in platinum-resistance emphasises the importance of the PI3K/AKT/mTOR signalling cascade as therapeutic target. Therefore inhibition of PI3K/AKT/mTOR signalling in ovarian carcinomas seems to be a promising target to enhance the efficacy of anticancer agents such as cisplatinum and to overcome the resistance of tumour cells against therapy. This hypothesis was tested in different preclinical in-vitro studies. Cancer cell lines are frequently used as invitro tumour models especially for analyzing and study of the effects related to a single gene. Today approximately 100 ovarian cancer cell lines are publicly available [93]. Some of these cell lines are known to be platinum resistant e.g. SKOV-3/DDP and Caov-3. Among the huge variety of ovarian cancer cell lines there are also the parental A2780 cells and the cisplatinum-resistant A2780cis cells [94]. Both cell lines are p53 and K-Ras wild-type and they have the same genetic background. The cisplatinum-resistant A2780cis cell line has been developed by chronic exposure of the parental cisplatinum-sensitive A2780 cell line to increasing concentrations of the chemotherapeutic agent [94]. These cell lines are excellent models for analyzing the molecular basis for cisplatinum resistance in ovarian cancer [72-74,95-98]. According to these studies AKT over-expression in ovarian cancer is at least one important reason for platinum resistance in this tumour entity [61,72,99]. It was shown that AKT protein expression is strong increased in cisplatinum-resistant A2780cis cell line compared to the parental A2780 cell lines [72,73]. The platinum resistance in A2780cis cell line could be overcome by AKT down-regulation via siRNA [72]. This was demonstrated in several functional in-vitro assays, e.g. clonogenicity assays, irradiation assays, determination of the apoptosis rate, and the cytotoxicity of cisplatinum was addressed in proliferation assays. Stable increase of AKT amount (AKT+) in the cell lines results in an increased IC50 value for cisplatinum whereas a stable decrease of AKT (AKT-) results in an increased accessibility for cisplatinum treatment [72] (table 3). Taken together it was shown in two cell lines with the same genetic background that AKT-overexpression rendered platinum-sensitive A2780 cells platinumresistant. Still more importantly, platinum-resistance of A2780cis cells could be reversed by down-regulation of AKT [72]. FACS analysis demonstrated that cisplatinum induces cell cycle arrest predominantly in the S and the G2/M phase but also in the G1 phase regardless of the AKT-expression status. However, required doses of cisplatinum were substantially higher in cell lines with AKT-over-expression [72,100].
As already mentioned above the sensitivity of cells to radiation and drug-induced apoptosis is determined by the balance between proapoptotic and anti-apoptotic protein expression [4,14]. Therefore the effect of the PI3K/AKT cascade on proapoptotic protein like BAD, a known substrate of AKT, has been studied in both cisplatinum-resistant Caov-3 and -sensitive A2780 human ovarian cancer cells [101]. Treatment of Caov-3 and A2780 cells with cisplatinum stimulated the activation of AKT, and the PI3K inhibitor wortmannin blocked the cisplatinum-induced AKT-activation. Cisplatinum treatment also stimulated phosphorylation of BAD at the Ser-112 and Ser-136 sites in Caov-3 and A2780 cells. Whereas phosphorylation of BAD at Ser- 136 was blocked by treatment with wortmannin, its phosphorylation at Ser-112 was blocked by a MAP/ERK kinase inhibitor, PD98059 [102]. Exogenous transient expression of a dominant-negative AKT in both Caov-3 and A2780 cells decreased cell viability after treatment with cisplatinum. In contrast, no sensitization to cisplatinum was observed in cells expressing wild-type AKT. These findings suggested that cisplatinum-induced DNA damage causes the phosphorylation of BAD via extracellular signal-regulated protein kinase (ERK) cascade and via a PI3K/AKT/mTOR cascade. Inhibition of either of these cascades sensitizes ovarian cancer cells to cisplatinum, thus providing further evidence that the AKT-pathway is involved in cisplatinumresistance in ovarian cancers [101]. Additional results suggest that AKT confers platinum-resistance, in part, by modulating the direction of p53 on the caspase-dependent mitochondrial death pathway [103]. Thus, in ovarian cancers p53 is a determinant of platinum sensitivity and AKT contributes to chemoresistance, in part, by attenuating p53- mediated PUMA upregulation and phosphorylation of p53 [104]. Recent results suggest that in platinum sensitive ovarian cancer cells, cisplatinum-induced apoptosis can also proceed, in part, via a caspase-independent mechanism involving apoptosis inducing factor (AIF), and that AKT activation additionally confers resistance to cisplatinum-induced apoptosis by blocking this pathway [103].
The anti-tumour effect of tangeretin, a citrus flavonoid known to inhibit cancer cell proliferation, was investigated in combination with cisplatinum in in-vitro models of A2780/CP70 and 2008/C13 cisplatinum-resistant human ovarian cancers [105]. Pretreatment of cells with tangeretin before cisplatinum treatment synergistically inhibited cancer cell proliferation. Interestingly, phospho-AKT and its downstream substrates, e.g., NF-κB, phospho-GSK-3β, and phospho- BAD, were down-regulated upon tangeretin-cisplatinum-treatment. The tangeretin-cisplatinum-induced apoptosis in A2780/CP70 cells was increased by PI3K inhibition and siRNA-mediated AKT silencing, but reduced by over-expression of constitutively activated AKT [105]. Although the overall results can only be interpreted with caution, as natural compounds such as tangeretin may display different effects apart from AKT-inhbition, tangeretin exposure preconditions cisplatinum-resistant human ovarian cancer cells for cisplatinum induced cell death. This effect may occur through downregulation of the PI3K/AKT/mTOR signalling pathway. In addition to these in-vitro studies, in a series of 98 patients with amplification of PI3K, an upstream component of the AKT-pathway was associated with resistance to platinum-based chemotherapy [106]. Accordingly, Woenckhaus et al. found that PIK3CA amplification was a strong predictor for early tumour associated death in ovarian cancer patients [107].
Recent work evaluated the anti-tumour efficacy of the AKT inhibitor perifosine in platinum-sensitive and –resistant human ovarian cancer cells [70,108]. In different ovarian cancer cell lines and in-vivo experiments it was possible to show that cells with higher levels of phospho-AKT are more sensitive to treatment with AKT-inhibitor perifosin. Furthermore, coincubation with perifosine sensitized A2780cis cells to treatment with cisplatinum. AKTinhibitor perifosine has already been tested in phase II studies in patients with breast, prostate, pancreatic, head and neck cancer, malignant melanoma, multiple myeloma, colorectal cancer and soft tissue sarcoma [109-115]. A recent phase I study with perifosine combined with radiotherapy performed in patients with advanced solid tumours has shown preliminary evidence of anti-cancer activity, including complete responses [116]. Thus, perifosine seems to be an attractive compound for further clinical studies in tumour entities, such as platinum-resistant ovarian cancers.
6. Role of AKT Expression Level in Tumour Cells in Regard to NK Killing
Another aspect in tumour diseases is the role of the immune system. As survival is strongly influenced by immunological parameters, immunotherapeutic strategies appear promising and during the last years the interest in tumour immunology has increased. A necessary prerequisite for immunotherapy in patients is a better understanding of the interaction between ovarian tumour cells and cells of the immune system especially natural-killer (NK)-cells. NK-cells are a critical component of the innate immune response against infectious pathogens and malignant transformation [117,118]. NK-cells mediate there activity through the elaboration of various cytokines as well as through direct cytolytic activity. However, unlike adaptive immune cells, which utilize specific clonal recognition receptors, NK-cell activation depends on a complex balance between activating and inhibitory signals [119,120]. NK-cells play an important role in immune surveillance and coordinating responses of other immune cells. Most tumour cells express surface molecules that can be recognized by activating receptors on NK-cells [121]. The expression of these receptors make such cells susceptible to endogenous NKcells, but malignant cells have developed mechanisms to evade these mechanisms of innate immune surveillance [122-124]. In patients with cancer, it is presumed that tumour cells have developed mechanisms to suppress NK-cell activation and resist lysis by endogenous NK-cells, but the molecular basis for target resistance is not well understood.
AKT seems to play also an important role in immune modulation. Recent studies have confirmed that AKT can regulate the development and functions of innate immune cells [125]. In this review only the role of activated AKT in tumour cells in regard to NK-cells will be addressed.
The PI3K/AKT/mTOR pathway regulates multiple cellular processes which underlie immune responses against pathogens or malignant cells [126,127]. Conversely, there is accumulating evidence that the PI3K/AKT/mTOR pathway is involved in the development of several malignant traits of cancer cells as well as their escape from immunity [128]. In some studies the interactions between cancer cells and natural-killer (NK)-cells have been enlightened [73,95,129-131]. Modified FATAL assay was used for determining the killing efficiency of NK-cells in regard to ovarian cancer cell models in-vitro. Therefore parental A2780 cells and the cisplatinum resistant A2780cis human ovarian cancer cells have been used. The efficiency of NK-cell mediated cell lysis differs between A2780 cells and the cisplatinum-resistant A2780cis cells. A2780cis cells are less accessible for NK-cell mediated killing [73,95]. This finding is in agreement with a report by Bellucci et al., using a lentiviral shRNA library targeting >1,000 human genes they identified 83 genes that promote target cell resistance to human NKcell- mediated killing [132]. Many of the genes identified in this genetic screen belong to common signalling pathways including members of the AKT/PI3K/mTOR pathway such as PIK3CA and PIK3CB [132]. The comparison of cancer cell lines A2780 and A2780cis revealed that the observed differences with regard to NK-cell mediated killing are most probably based on two mechanisms. First of all the observed increased expression of anti-apoptotic genes (especially ciap-1 and -2) in A2780cis cells compared to A2780 cells most probably renders A2780cis cells more resistant against apoptosis. Second the CD112 ligand for NK-cell receptor DNAM-1 was expressed at a reduced level in A2780cis cells but ligands for the NK-cell receptor NKG2D, e.g. MICA/B, were expressed more strongly in the platinum-resistant cells compared to parental A2780 cells [73]. A2780cis cells express lower levels of TIMP-3 the inhibitor of MICA/B shedding. At the same time the proteases for shedding are expressed which result in a net increase of soluble MICA/B in A2780cis cell cultures [73]. It is well known that cleaved MICA/B protect cells against NK mediated cell killing [73,133,134]. Therefore, it is very likely that most probably the increased amount of soluble MICA/B is responsible for the lower killing rate of platinum-resistant A2780cis cells compared to their parental A2780 cells [73]. Previously it was demonstrated that PI3K/ AKT/mTOR pathway is involved in inducing MICA/B expression in breast cancer cells [135]. This finding seems to be a more general effect of an induced PI3K/AKT/mTOR signal transduction way; also in ovarian cancer cells with an increase in phosphorylated AKT/ activated PI3K/AKT/mTOR pathway higher MICA/B expression has been detected [73]. Recently it was demonstrated that treatment of tumour cells with JAK inhibitors increased their susceptibility to NK-cell mediated killing [132]. The authors concluded that common signalling pathways can regulate susceptibility of human tumour cells to killing by immunologic effector cells and that small molecule inhibitors of these kinases may have important immunologic effects in-vivo [132]. Whether in analogy inhibition of PI3K/AKT/mTOR pathway renders the platinum-resistant A2780cis cells accessible for NK-cell mediated killing must be evaluated in further studies. Only the first steps towards the characterization of the molecular basis for resistance mechanisms in ovarian cancer with different AKT expression levels regarding NK-cell mediated killing are done [73,95].
7. Conclusion
Alterations of the serine/threonine kinase AKT/PKB pathway have been detected in several human malignancies including ovarian cancer [136]. AKT has a broad range of downstream effectors that regulate cell processes such as cell growth, cell cycle progression, survival, migration, epithelial-mesenchymal-transformation and angiogenesis [76]. Furthermore AKT seems to be able to influence directly the immune resistance of malignant cells e.g. ovarian tumour cells [73,95,129-131]. Comparison of parental and immuneresistant ovarian tumours revealed that AKT is highly activated in the immune-resistant tumours. The observed resistance against apoptosis was found to be associated with the up-regulation of anti-apoptotic molecules [73].
The AKT pathway is a promising target for cancer therapy, as it is a main nodal point where extracellular and intracellular oncogenic signals are integrated. Due to the key role of AKT in malignant transformation numerous inhibitors of the AKT-pathway have been developed, and are currently in various stages of clinical development [32]. In human specimens of ovarian cancer AKT was found to be activated in 68% [136] and PI3K, an upstream component of the AKT-pathway, was found to be mutated in 12% of the cases [64]. Recent evidence has shown that overactivation of the PI3K/AKT/mTOR-pathway may be associated with platinum resistance [20,70-72,78,95]. It was demonstrated that parental A2780 cells become platinum resistant by over-expression of AKT and that platinum resistance in A2780cis cells can be overcome by transfection with siRNA down-regulating AKT [72].
Due to different deregulation that can occur in the PI3K/AKT/ mTOR pathway, as outlined above, there will exist in tumour patients most probably differences that should be taken in account and treated in different ways. That is a strong argument for the need of patient orientated medicine, the so called personalized medicine. In the light of modern very sensitive methods like proteomics, nextgeneration- sequencing, digital-droplet-PCR and array technologies like NanoString it should be possible to test every tumour patient for the presence or absence of relevant mutations as well as miRNA and protein expression pattern to come up with the best possible individual treatment for each tumour patient.
Even if we have today a huge knowledge about signal transduction, regulation and interaction of different signal transduction pathways it seems that there still is at least partially lack in understanding the complex network of protein interaction in detail. Therefore further research is necessary to unravel all protein interactions and especially in the case of PI3K/AKT/mTOR pathway the effects of protein overexpression in the context of different cells and patients.
Competing Interests
The authors declare that they have no competing interests.