


1. Introduction
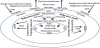
Unicellular microorganisms are able to develop multicellular and structured communities, a phenomenon termed biofilms, discovered in the latter half of last century [1]. Biofilms, in fact, predominate in nearly all nutrient-sufficient ecosystems [2], natural and clinical. Unlike the bacteria that inhabit natural environments, bacteria that live on biotic matter, e.g., human hosts, or abiotic environments, such as hospital settings, often encounter antimicrobial and DNA damaging agents. The understanding that biofilms contribute to antimicrobial resistance [3,4] led to a thought that such agents at sub-inhibitory concentrations might induce biofilm formation [5]. This hypothesis has been tested for a decade. The relevant findings come from a wealth of studies. For example, biofilm formation of Pseudomonas aeruginosa and Escherichia coli was induced by the sub-inhibitory aminoglycoside antibiotics [6,7]. Vibrio cholerae biofilm formation was increased by bile acids [8]. Biofilm formation of Mycobacterium avium was stimulated by hydrogen peroxide [9]. Further, P. aeruginosa biofilm formation was stimulated by DNA replication inhibitors and the antibiotics, such as quinolone antibiotics, hydroxyurea, nalidixic acid and ciprofloxcian [10-14]. These observations demonstrate that biofilms are induced when the bacteria encounter antimicrobial stress, so termed the stress-inducible biofilm [12]. Because its formation is induced by antimicrobial treatment, such biofilm must have a profound impact not only on clinical practice of antimicrobial treatment, but also on the molecular evolution of antimicrobial resistance in biofilms. As more and more antimicrobials have been found to trigger the SOS response of bacteria (Figure 1) [15], some of which increase biofilms, this article intends to review the research progress about the link between biofilms and SOS of bacteria.

2. The SOS Response to Antimicrobials Induces Biofilm Formation
Bacteria launch an emergency response, named after the international telegraph distress signal termed “SOS”, when they encounter environmental threats, such as antimicrobials and UV radiation. The original hypothesis for SOS came from the observation that reactivation of the UV-irradiated phage increased greatly when the viruses replicated in irradiated E. coli cells [16], a phenomenon so called Weigle reactivation [17]. Further, when E. coli lysogens that bored prophage λ in their chromosome were irradiated by UV, prophage was induced, causing cell lysis and phage release [18-21]. The cell division was inhibited. The aftermath of radiation including division arrest, prophage induction and UV-induced mutation, was thought to be interrelated [22] and collectively concluded as DNA damage repair through the SOS response (Figure 1) [17,23]. The SOS response, in particular, is controlled by interplay of an SOS repressor LexA (locus for X-ray sensitivity A [24]) and a sensor/inducer RecA (recombinase A). They alternatively switch SOS on or off (Figure 1) [25] as reviewed previously [26]. In the off-mode, LexA represses expression of the SOS genes via binding to the conserved sites (SOS boxes) upstream of these genes in the absence of significant DNA damage (Figure 1). There are at least 43 SOS genes mostly involved in DNA damage repair in E. coli [27,28]. In the on-mode, RecA, a coprotease, senses DNA damage signals [single-stranded (ssDNA)] and becomes activated to assume a filament structure able to bind to ssDNA [29,30]. The ssDNA-RecA coprotease complex activates autocleavage activity of the LexA polypeptide (Figure 1) [25]. Once self-cleaved, LexA dissociates from the SOS boxes upstream of the SOS genes and derepresses these genes. SOS is thereby turned on. The phage λCI repressor is also auto-cleaved, resulting in prophage induction (Figure 1) [31]. The SOS gene products of the bacterial host act to repair or bypass DNA damage, but those of phage λ switch the viral life style from lysogenic to lytic growth (Figure 1) [32]. With a bacterial cell recovering from the DNA damage and the activity of RecA coprotease declining, LexA becomes dimmerized and turns off SOS by binding back to the SOS boxes [33].
The link of the SOS response to biofilm formation was implicated by the finding of the dual effects of some DNA damaging agents on SOS and biofilm inductions [11]. These antimicrobial agents at the sub-inhibitory concentrations, such as hydroxyurea, nalidixic acid and ciprofloxacin, induce the SOS response in most bacteria [15], and this response insures cell survival. Surprisingly, these agents stimulate biofilm formation [10-14]. Additionally, hydrogen peroxide that triggers oxidative stress and the SOS response [34] stimulates biofilm formation in M. avium [9]. To study the role of SOS in biofilm formation, we can lock the SOS circuit in the off mode even in the presence of the SOS signal (Figure 1). This has been achieved by introducing mutations that block the self-cleavage of LexA so that the mutant LexA protein becomes non-cleavable and the lexA Ind(-) mutant becomes non-inducible, repressing the SOS genes even when the cells encounter DNA damage [35]. With the lexA Ind(-) mutant, the effects of SOS on biofilm formation can be measured by comparison of biofilm masses between the wild-type strain and the mutant in the presence of DNA damage agents [36]. Alternatively, a recombinase A deficiency by null mutations can preclude the SOS response [37]. The pleotropic effects the recA mutations may complicate the experimental analysis in some cases.
With construction of the SOS mutants, great progress has been made in testing the connection of SOS with biofilms (Table 1). The recombinase A deficiency of Streptococcus mutans that precludes the SOS response reduced not just acid tolerance but density and cellular viability of biofilm [38]. Fibronectin-binding proteins (FnBPs) of Staphylococcus aureus, the components of a proteinaceous biofilm, was induced by activation of a LexA-dependent SOS response [39]. Repression of the SOS regulons in E. coli with engineered bacteriophage to over-express lexA increased not only bactericidal effects of antibiotic-resistant bacteria, persister cells, and biofilm cells, but reduced the number of antibiotic-resistant bacteria arising from an antibiotic-treated population [40]. Listeria monocytogenes formed biofilms under continuous-flow conditions, under which the SOS response was induced. Deletion of yneA, the SOS regulon member, reduced the biofilm formation [41]. The most conclusive experiments at that time were performed [12] with P. aeruginosa SOS strains carrying mutations inactivating SOS sensor RecA [37] or rendering LexA repressor non-cleavable [35]. The results indicated that biofilm production was stimulated by a DNA replication inhibitor hydroxyurea at the sub-inhibitory concentration, and the stimulation diminished by either the recA knock-out mutation in which SOS was precluded or the non-cleavable LexA mutation in which SOS was not inducible [lexA Ind(-)][12]. Evidently, as biofilm formation decreases when SOS is in the off-mode, these findings support a conclusion that SOS contributes to biofilm formation. This conclusion was further tested. Treatment of P. aeruginosa with DNA-damaging antibiotic ciprofloxacin at sub-inhibitory concentrations also stimulated biofilm formation [14]. The biofilm mass was reduced when SOS was noninduced in the lexA Ind(-) mutant, the results indicative of SOS involvement in biofilm formation. The decrease in biofilm appears due to LexA repression of motility, which was involved in the initial adherent event in biofilm development [14]. These results suggest that bacterial adhesion increases during SOS. This conclusion receives further support from a study of adherence of E. coli K-12 strains to abiotic surface. The attachment, in fact, induced the SOS response that subsequently enhanced biofilm formation since the lexA Ind(-) mutant did not form biofilms [42]. Thus, bacterial adhesion mediated by flagella appears to increase under SOS (Figure 2).


3. SOS Plays a Role in Biofilm Maturation
Not just the initial adhesion, the downstream events are also affected, such as biofilm maturation, cell death, dispersal and variant formation in the biofilm life cycle. Of those, the biofilm variant formation relies on a filamentous Pf4 prophage [43] that was associated with cell lysis and DNA release into biofilm matrix. Although the Pf4 phage was partly controlled by two-component regulator BfmR through PhdA [44], conversion of the Pf4 phage into a superinfection (SI) variant was activated by DNA damage and the resulting SOS response (Figure 2) [45]. As SI variants reinfect and kill the prophage-bearing hosts in biofilms, DNA released from the dead cells is likely to go into biofilm matrix for biofilm maturation as reviewed recently [46,47]. Understandably, when the bacteria that are lysogenized by phage DMS3 acquire immunity to the phage infection, both biofilm formation and swarming motility are inhibited [48,49]. Such an adaptive immune system present in many archaea and bacteria is operated by clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated (Cas) proteins in which a small portion of viral DNA is inserted at the CRISPR locus in the host genome to provide adaptive immunity to the mobile genetic element [50,51]. One of possible mechanisms for the CRISPR-dependent inhibition of biofilm formation is disruption of cell adhesion in that the attached cells are killed due to inability of the CRISPR-engaged strain to downregulate expression of the SOSregulated phage-related genes (Figure 2). The inhibition is biofilmspecific as growth and viability under planktonic conditions are not noticeably affected by these phage-related genes [52]. Since bacterial attachment activates the SOS response [42] and phage-related gene expression [52], it seems paradoxical that SOS enhances biofilm maturation while inducing suicidal expression of phage-related gene of the attached cells. Although biofilm formation by lysogens of a single species is inhibited by attachment-activated SOS and inability of CRISPR to the subsequent expression of phage-related genes, biofilms of mixed strains of lysogens and non-lysogens are likely to increase via several mechanisms, one of which is DNA release from lysed lysogens into biofilm matrix (Figure 2). Based on our current understanding, SOS plays multiple roles in biofilm development starting from adhesion to maturation via the phage-mediated matrix DNA release and genetic variation. Yet, the role of CRISPR in maturation of heterogeneous lysogens-nonlysogen or multi-species biofilms needs further study.
4. SOS Contributes to Biofilm Defense Against Stress
Bacteria form biofilms in many habitats, either natural or experimental, by attaching to either abiotic or biotic surfaces, to protect themselves from environmental hazards [1]. Biofilms can be environmental reservoirs of antibiotic resistance as reviewed recently [53]. The biofilm matrix of P. aeruginosa displays protective properties against UV radiation [54]. Biofilms cells are 10-to-1,000 folds less susceptible to certain antimicrobial agents than their planktonic counterparts [55]. Obviously, biofilms are defensive against hazardous agents. There are several factors that contribute to the biofilm defence as summarized lately [53], such as the barrier of polysaccharide matrix, the slow or no growth of some biofilm cells, the resistant phenotype of persister cells, and the genotype of antibiotic resistance genes. SOS plays an important role in the defence mechanisms. First, SOS is implicated in biofilm resistance to antimicrobials due to antibiotic resistance genes in bacterial genomes or plasmids (Figure 3). One of these genes encodes β-lactamase that inactivates the β-lactam antibiotics. The β-lactamase-producing bacteria increase the protection in biofilms [56]. These resistance genes are often carried by plasmids, of which the transfer rate can be enhanced by sub-inhibitory concentrations of tetracycline and cephradine among the biofilm biomass [57]. The β-lactam antibiotics induce SOS [58], under which both the copy number of resistant plasmids and the production of β-lactamase are increased [59]. The horizontal transfer of virulence factors is also activated by SOS [60]. Moreover, β-lactamases are encoded by the integron-borne genes [61], and the gene rearrangement is mediated by the integrase IntI1 [61,62]. Its expression is SOS-induced in vitro [63]. This SOSregulated expression plays an important role in clinical acquisition of antibiotic resistance [64]. Blockage antibiotic-induced activation of the SOS response by RecA inhibitors potentiates bactericidal activity of antibiotics and hinders bacterial acquisition of antibiotic resistance mutations and horizontal transfer of resistance [65]. Obviously, the biofilm resistance mediated by the resistance genotypes via vertical and horizontal transfers can be attributed to SOS; yet direct connection to biofilm defence remains to be established.

Second, the tolerance and resistance to antimicrobials are facilitated in biofilms. In particular, some subpopulations highly tolerant to the stressors in biofilms are likely to be generated and selected during SOS. This premise is aligned with the following findings. Treatment of E. coli with ciprofloxacin, a DNA-damaging and SOS-inducing antibiotic, caused formation of persisters [66], which are dormant cells highly tolerant to antibiotics and involved in recalcitrance of biofilm infections (Figure 3)[67,68]. Persister formation was induced by some components of the SOS response after exposure to a DNAdamaging antibiotic [69]. Isolated persisters displayed an increase in the levels of toxin/antitoxin (TA) transcripts, and SOS induced many TA genes; but knocking out a SOS-TA locus, tisAB/istR, reduced the level of persisters tolerant to ciprofloxacin. Thus, a SOS-induced toxin possibly controlled persister formation [66,70]. Third, the foodborne pathogen Listeria monocytogenes, able to form biofilms in the food processing environment, generated genetic variants specifically induced in continuous-flow biofilms but not in static biofilms. The generation was dependent on radical-induced DNA damage and RecA-mediated repair [71]. Lastly, the starved biofilms of E. coli displayed higher tolerance towards fluoroquinolone ofloxacin than their planktonic counterparts (Figure 3). The biofilm and starvationassociated tolerance depended on biofilm age and SOS. The tolerance did not involve the SOS-induced toxin-antitoxin systems related to formation of highly tolerant persisters. These results demonstrate that SOS is induced in the nutrient-deprived biofilm microenvironments is the mechanism by which biofilm-specific tolerance to the fluoroquinolone ofloxacin develops (Figure 3) [72]. Altogether, SOS appears responsible for generation of persisters, genetic variants and starvation-associated tolerance in biofilms treated with antimicrobial agents, which in turn select for the generation outcomes so that evolution is likely to occur.
5. Biofilms as Hotspot of Evolution Driven by SOS
Biofilms, either experimental or nature-occurring, provide niches structurally, biochemically and genetically for bacteria to evolve as reviewed recently [73,74]. Bacteria, albeit unicellular, form multicellular communities shielded by a polymer matrix in biofilms [2,75,76]. Transportation through these enclosed communities is uneven, responsible for formation of gradients of both nutrients and toxic materials (Figure 4). This eventually leads to a spatially structured, heterogeneous environment inside biofilms [77,78]. Such an environment facilitates diversification within biofilms. Biofilms are therefore the hotspot of natural selection that can be fueled by antimicrobials and the SOS response (Figure 4) as SOS is involved in antibiotic resistance evolution [40,79] and in regulation of evolvability [80,81]. For instance, almost all bacteria produce bacteriocins, the antimicrobial compounds that are usually released through lysis of the producer strains under environmental stress [82]. For the toxic effect on the neighboring cells, bacteriocin production is under tight regulation, especially the SOS control [83]. Some of bacteriocins are biofilm-related. The biofilms formed by a strain of the natural isolate E. coli specifically produce a new pore-forming colicin R, which is not obviously present in the planktonic culture [84]. The colicin R expression is SOS-dependent since the SOS response that is activated within mature biofilms induces the expression. Surprisingly, colicin R is toxic to biofilm cells but not to planktonic cells. In the mixed biofilms, this biofilm toxicity provides the colicin-producing bacteria with a competitive advantage by outcompeting susceptible neighboring bacteria [84]. Additionally, P. aeruginosa produces pyocins, whose expression is repressed by PrtR. This negative regulator is cleaved under the SOS response, leading to derepression of pyocin genes [83]. Accordingly, hydrogen peroxide or ciprofloxacin known to induce the SOS response increases pyocin production [85]. The treatment also elevates the PrtR mRNA level due to cleavage of the autorepressor PrtR for its own gene [85]. Since SOS is induced in biofilms, pyocin activity is increased in biofilms, especially the slow-growing anaerobic population [86]. In mature biofilms made of aerobic and anaerobic communities, pyocins impose a significant impact on biofilm population dynamics (Figure 4).
With the stress-inducible biofilms produced under SOS and selection of antimicrobial agents, the biofilms may be the hotspot where microevolution inevitably occurs. Nevertheless, the cells in the biofilms seem able to differentiate and evolve into purposeful structures for offense and defense (Figure 4). Of the multiple-species biofilms in soil, the antibiotic pyocyanin-producing bacteria of one species coexist with the pyocyanin-sensitive bacteria of another [87]. How the vulnerable live with their killers inside biofilms is very interesting. It was found that a layer of pyocyanin resistant bacteria evolved between the pyocyanin-producing and pyocyanin-sensitive bacteria so that the susceptible were protected from their killers [87]. This finding indicates that biofilms are not just of genetic diversity but of acquiring the capacity to evolve a highly organized society of defense and offense (Figure 4). This social architecture was even emulated by an 'onion model' in which phenotypic diversification was produced in biofilms [88]. Elevated phenotypic variance among the onion-like layers increased likelihood of resistant subpopulations emerging to selective agents [88]. In particular, one of the selective agents intrinsic to biofilms is oxidative stress under which hydrogen peroxide triggers the SOS response (Figure 4) [34]. The biofilms with the onion-like architecture was, in fact, prone to the hydrogen peroxide-mediated oxidative stress that often induced SOS [34] and resistant mutability to antibiotics [89]. Since DNA replication and repair under SOS are error-prone and mediated somewhat by SOSregulated DNA motor proteins [90,91], mutability is likely to rise up. The mutability augmented via SOS can impair functions of the genes for the mismatch repair system (MRS), leading to MRS deficiency. This can leave numerous mismatch errors in DNA unrepaired and generate mutations. The MRS deficiency and the subsequent increases in mutation rates were found to drive the increased phenotypic diversification of P. aeruginosa biofilms (Figure 4) [89] and evolution of antibiotic resistance during development of E. coli biofilms even in the absence of antibiotic selection [92]. Therefore, biofilms are like a brewing fortress where the SOS response to external and internal stresses fuels mutagenesis that promotes phenotypic diversification of the members to form defensive zones (Figure 4). Biofilms appear to be a hotspot of bacterial evolution that is driven by the SOS response.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
Weitao contributes to conception, design and writing of this article.
Leiker to revising it critically for important intellectual content.
Acknowledgments
We thank Karin Sauer for insightful comments.