


1. Introduction
Langerhans Cells (LC) is a subset of antigen-presenting dendritic cells belonging to the histiocytic system. The World Health Organization (WHO) classifies LC tumors into Langerhans cell histiocytosis (LCH) and Langerhans cell sarcoma (LCS). LCS is a rare neoplastic proliferation of LC with overtly malignant cytologic features. The condition can present de novo, or it can derive from a previous LCH [1,2]. Histiocyte tumors are among the more uncommon neoplasms affecting lymphoid tissues, probably representing less than 1% of all malignancies involving lymph nodes or soft tissues [3-4]. They can occur at any age, although incidence peaks around the fourth decade of life. Skin and the underlying soft tissues are the most common sites of tumor origin, but the involvement of bone, lung, gallbladder, and peritoneal lymph nodes has also been described [5-7,1].
The diagnosis of cutaneous LCS can be difficult: metastatic cancer, malignant melanoma, anaplastic large-cell lymphoma, and myeloid sarcoma show similar microscopic features. LCS can be limited to the skin, or it can spread to other organs [8]. To the best of our knowledge, primary cutaneous LCS without any extracutaneous associations is extremely rare; we describe a young male patient with this rare condition.
2. Case Report
A 25 year-old Caucasian male, otherwise healthy, was referred to a dermatologist for a single rapidly growing lesion on his left leg. The 12-mm nodule was firm and erythematous, with central ulceration (Figure 1). He reported no weight loss and there was no palpable lymphadenopathy or organomegaly. Surgical resection of the skin lesion was performed without preoperative skin biopsy.
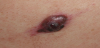
The surgical specimen was noted to be a subcutaneous nodule, yellowish in color. The nodule was fixed in 10% neutral buffered formalin and then embedded in paraffin and sectioned in 4-μmthick slices. The slices were then stained with hematoxylin and eosin. Immunohistochemical analyses were performed using the ChemMate Envision/HRP Kit (Dako, Glostrup, Denmark). Antibodies against vimentin, S-100 protein, cluster of differentiation CD 68, CD1a, langerin, pan-cytokeratin, melan-A, and Ki-67 were obtained from DakoCytomation (Dako, Glostrup, Denmark).
3. Pathological Findings
The irregularly shaped nodule showed a central ulceration, with diffuse infiltration of large tumor cells into the dermis. The tumor cells had significant malignant cytological features: they exhibited an irregular shape with abundant eosinophilic, sometimes clear, cytoplasm and large, irregular shaped nuclei with a lobulated or dented appearance. Some cells displayed a longitudinal nuclear groove and prominent nucleoli. A high mitotic rate (more than 30 mitoses per 10 high-power fields) was observed. Moderate inflammatory infiltration consisting of neutrophils, plasma cells, eosinophils, lymphocytes, and macrophages was observed, interspersed with the tumor cells (Figure 2).

4. Immunohistochemical Staining and Results
Immunohistochemistry staining showed that the tumor cells were diffusely positive for vimentin, S-100, CD68, CD1a, and langerin but negative for melan-A and pan-cytokeratin. The Ki-67 proliferating index was high at approximately 12%.
The nodular growth pattern, significantly malignant cytological features, high mitotic index, and immunohistochemical aspects led us to the diagnosis of a cutaneous LCS, according to the WHO diagnostic criteria (Figure 3, 4).


5. Discussion
Histiocytoses are rare neoplasms with a highly unpredictable clinical course ranging from spontaneous regression to highly aggressive, even fatal, disease. LCS has the potential to progress to leukemia [3,8]. The typical sites for LCS development are skin, bone, and lymphoid tissues. The diagnostic criteria of LCS include the presence of cells that have both characteristics of LC and typical histological features of tumor cells. The typical immunophenotype features langerin, CD1a, and S-100 are helpful to achieve an accurate diagnosis of these dendriticcell tumors. Nonetheless, the diagnosis of LCS can be difficult: tumor cells with poorly differentiated or atypical morphology might occasionally lose the distinctive immunophenotype.
According to the WHO classification system, tumors of the dendritic-cell lineage include follicular dendritic-cell tumors, interdigitating dendritic-cell tumors, LC tumors, and other rare dendritic-cell tumors. LC and interdigitating dendritic cells share a common hematopoietic CD34+ precursor; in contrast, follicular dendritic cells do not have a hematopoietic origin. LC tumors show expression of both CD1a and S-100 protein, while interdigitating cells are positive for S-100 but negative for CD1a. Follicular dendritic cells express CD21 consistently, but never express CD1a [9].
Cutaneous LCS, with its rareness in skin and its poorly differentiated morphologic features, shares a differential diagnosis with other epithelial or mesenchymal skin neoplasms. All of these diseases exhibit skin lesions and a frankly malignant cytologic appearance with a highly aggressive clinical course and a poor prognosis. However, cutaneous squamous-cell carcinoma and metastatic cancer both display an obvious nest structure with an epithelial phenotype including pan-cytokeratin, cytokeratin CK 7, or CK20. Melanoma might share S-100 protein positivity with LCS, but it also expresses other melanocytic markers such as human melanoma black (HMB)45 and melan-A. Anaplastic large-cell lymphomas are positive for CD30 and epithelial membrane antigen (EMA) and may show positivity for anaplastic lymphoma kinase (ALK). Myeloid leukemia can originate in the skin, and CD68 and lysozyme positivity may be observed, which could make this condition difficult to distinguish from LCS. The presence of myeloid-specific markers, such as myeloperoxidase (MPO), CD117, CD99, and CD34, should be helpful in differentiating cutaneous myeloid leukemia from cutaneous myeloid sarcoma [10]. In our patient, the tumor cells strongly expressed CD1a, S-100 protein, and langerin, markers specific for LC. These findings argued against other cutaneous hematological neoplasms.
6. Conclusion
In conclusion, LCS is extremely rare and only a few cases of LCSs with exclusive cutaneous involvement have been reported in the literature. Our patient represents this rare category of disease. The diagnosis of primary cutaneous LCS is difficult and should be made cautiously, particularly in patients with clinically localized disease who lack extracutaneous manifestations.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
Maurizio Giuliani: wrote the paper, had drawn the study and gave
the final approval.
Alessandro Marinucci: performed the laboratory analysis, had
cooperated in study drawing.
Loredana Melchiorri: performed the laboratory analysis, had
cooperated in study drawing.
Giovanni Zoccali: performed the literature review and revisited the
manuscript.