


1. Introduction
Oculo-auriculo-vertebral spectrum is a term suggested by Gorlin to summarize the different phenotypic expressions of a continuum that has been known as hemifacial microsomia, Goldenhar syndrome, or first and second branchial arch anomalies with areported prevalence of up to 1/3500 births [1].
OAVS affects males more frequently than females by an approximate 3:2 ratio [2-4]. OAVS is aetiologically and pathogenetically heterogeneous. In most cases OAVS appears to occur randomly, with no apparent cause (sporadic). However, in some cases, family histories suggest autosomal dominant or recessive inheritance. The presence of familial cases, following autosomal dominant [5-8] or autosomal recessive inheritance [3,4,9]. As well as several chromosomal aberrations have been reported on previously in patients with OAVS and several candidate genes have been proposed but no one has been confirmed as causative of the phenotype [10-13]. In addition, some authors propose a multifactorial pattern of inheritance [14]. Environmental causes have also been suggested, such as maternal diabetes during pregnancy, gestational or pre-existing vasoactive drugs, smoking and twinning, assisted reproductive technology indicating that a multifactorial aetiology (environmental and genetic) also contributes to some cases of OAVS [15].
Due to the variable expressivity, there is no consensus regarding the minimum diagnostic criteria for OAVS. Tasse et al [8] suggested either isolated microtia or hemifacial microsomia together with mild ear malformations, such as preauricular tags and hillocks, (suggested to be variants of microtia) as minimal diagnostic criteria. Beleza- Meireles et al. [16] suggested that the presence of isolated hemifacial microsomia associated with a family history of OAVS should also be considered to be diagnostic.
Here, we report on four unrelated patients who have distinct phenotypic anomalies compatible with OAVS (Table 1).
2. Case Reports
2.1 Case 1
A 4-year and 8-month-old boy was admitted to Pediatric Genetics clinic with speech delay and left-sided anotia. He was born at 32th gestational wk, 1650 gr, by cesarean/section (preeclampsia). He was the only child of a non-consanguineous family. Her father had bilaterally anotia with congenital hearing loss. He sat without unsupported at 8th mo and able to walk at the age of 12 mo. He started to speak by the age of 2 but was not able to make sentences. He had right-sided inguinal hernia operation at 18th mo. He was followed with patent ductus arteriosus and atriel septal defect up to 3 yr from Pediatric Cardiology clinic. On his examination his weight was 15 kg (10th centile), height was 104 cm (25-50th centile) and frontooccipital head measurement was 47 cm (-2SD>). He had severely malformed peanut-shaped auricle with undeveloped external ear canal on the left-side, micrognathia and double urethral orifice (Figure 1). Hearing test, applied to the right ear, was normal. However the test could not apply to left side due to absence of left external ear canal. On his cranial magnetic resonance imaging, abdominal ultrasonografic screening and vertebral roentgenograms, no additional abnormalities were demonstrated. The Ankara Developmental Screening Inventory was applied to the patient and the psychomotor development of the patient was found normal. He was advised to take speech therapy.

2.2 Case 2
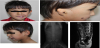
A 5-year and 4-month-old boy was referred to Pediatric Genetics clinic because of microtia and caudal regression syndrome. He was born at 38th gestational week, 2710 gr, by cesarean/section. He was the second child of a non-consanguineous family. Her mother was disagnosed as gestational diabetes mellitus and took insulin treatment through the third trimester. He had right-sided choanal atresia repairment on 65th days of his life. One year ago, he admitted to Pediatric Nephrology clinic with enuresis diurna and got diagnosis of caudal regression syndrome. He started to walk and speak at the age of 15 mo. On his examination his weight was 15 kg (3th centile), height was 106 cm (10th centile) and frontooccipital head measurement was 40 cm (-2SD>). He had right-sided peanut shape auricula with intact external ear canal, peripheric facial palsy and sacral dimple (Figure 2a, Figure 2b and Figure 2C). Vertebral roentgenogram and lumbosacral magnetic resonance imaging showed the agenesis of sacrum and coccyx (Figure 2d and Figure 2e). On his cranial magnetic resonance imaging, abdominal ultrasonografic and echocardiographic screening, no additional abnormalities were demonstrated. Hearing test was applied for both ear and evaluated as normal.
2.3 Case 3
A 2-year and 7-month-old boy was referred to Pediatric Genetics clinic because of unilateral faciel clefting and anotia. He was born as a second child of a non-consanguineous family at 40th gestational wk, 3600 gr, by spontaneous vaginal delivery. He failed to pass meconium within 48 hr of delivery and was diagnosed as Hirsprung disease on the postnatal 3rd day (Figure 3). He had cryptorchidism operation at the age of 8 mo and has undergone multiple surgeries with Hirsprung disease up to 2 yr. He sat without unsupported at 8th mo, able to walk at the age of 12 mo and started to speak by the age of 12 mo. On his examination his weight was 13 kg (25th centile), height was 92cm (50th centile) and frontooccipital head measurement was 45 cm (-2SD>). He had left-sided severely malformed auricle with absent external ear canal, Tessier 7 type orofacial clefting and mandibular hypoplasia (Figure 3). Hearing test, applied to the right ear, was normal. Echocardiograhic screening showed muscular ventricular septal defect. However the test could not apply to left side due to absence of left external ear canal. On his cranial magnetic resonance imaging, abdominal ultrasonografic screening and vertebral roentgenograms, no additional abnormalities were demonstrated.
2.4 Case 4
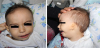
A 5-month-old boy was referred to Pediatric Genetics clinic because of his dysmorphic appearence. He was born as a second child of a non-consanguineous family (his mother and father were from same village) at 39th gestational wk, 2000 gr, by cesarean/section. It was learned that his first degree cousin (the son of his uncle) had unilateral facial microsomia. He could hold his head in first month, recognize and smile her mother for 4 mo. On his examination his weight was 4000 gr (3th centile >), height was 60 cm (3th centile) and frontooccipital head measurement was 38 cm (3th centile >). He had triangular face, downslanding palpebral fissures, arched eyebrows, broad and low nasal root, hypoplasia of the left ala nasi, thin upper lip, down turned mouth, mandibular hypoplasia, left-sided severely malformed auricle with intact external ear canal and bilateral preauricular skin tag (Figure 4). Hearing test, applied to the right and left ears, were normal. On his cranial magnetic resonance imaging, abdominal ultrasonografic screening and vertebral roentgenograms, no additional abnormalities were demonstrated.

3. Discussion
OAVS is an extremely complex and heterogeneous condition that affects primarily aural, oral and mandibular development. The disorder is characterized by a wide spectrum of symptoms and physical features that may vary greatly in range and severity from case to case [1,5].
The exact etiology is not known. However, it is possible that abnormal embryonic vascular supply, disrupted mesodermal migration or some other factor leads to defective formation of the branchial and vertebral systems [17].
Most cases of OAVS reported are sporadic but familial cases have been described. A few families were reported to have an autosomal recessive inheritance [3,4,9], other families’ presentation of the syndrome strongly supported an autosomal dominant inheritance [5-8]. Two of our four cases had a positive family history, one of them was represented an autosomal dominant (case 1), and the other was autosomal ressesive mode of inheritance (case 4).
OAVS is usually more common in male [2-4] and the male predominance in four cases was supported the literature.
Hemifacial microsomia is a common birth defect, typically affects the external ear, middle ear, mandible and temporomandibular joint, muscles of mastication and facial muscles, and other facial soft tissues on the affected side. Microtia with or without preauricular skin tags may represent the mildest form of the OAVS and has been considered as minimal diagnostic criteria for this condition [5,8]. Jin et al. [18] reported 28.4% of cases with microtia accompanied by hemifacial microsomia and preauriculer tags/sinusus in %14,4 and eksternal meatus stenosis/atresia in % 98,6. Hemifacial microsomia was accompanied to microtia in two cases (cases 3 and 4), eksternal meatus stenosus in three cases (cases 1,3 and 4) and only one of them had bilateral preauricular skin tag (case 4).
Facial asymmetry associated with CFM are more likely involve the right side of the face than the left side of the face [19]. However, all of the cases except case 2 had left-sided involvement.
Case 3 had orofacial clefting (Tessier no:7). Though clefts of the orofacial region are among the most common congenital facial defects, the occurrence of lateral facial clefts (Tessier 7 cleft) in conditions such as the OAVS, is very rare (<5%) [20].
Malformed and/or fused cervical vertebrae are common, though anomalies can be noted throughout the spine and hemivertebrae are common in OAVS [21]. Maternal diabetes is known to have teratogenic effects. OAVS occurs with a higher incidence in infants of diabetic mothers [22]. Only one in four cases had vertebral anomalies that this case (case 2) was an offspring of a diabetic mother and he had caudal regretion syndrome. He had also additional anomalies such as unilateral coanal atresia and facial paralysis.
Two in four cases had congenital cardiac defect (cases 1 and 3) Cardiovascular anomalies have also been associated with OAV [23]. The frequency of cardiovascular heart defects in OAV ranged from 5% to 58% with conotruncal defects and septal defects as the most common among patients, as it was manifested in our patient [24].
Limb, renal and central nervous system anomalies have also been observed in patients with OAVS [25]. Two of them had urogenital abnormalities. Case 1 had right-sided inguinal hernia and double urethral orifice. Case 3 had left sided cryptorchidism. None of them had central nervous system abnormalities. None of them had development delay nor intellectual disability. Only case 1 had delay of speech because of having deaf parents.
Only case 3 had gastrointestinal abnormality (Hirsprung disease). Gastrointestinal system abnormalities accompanied to OAVS was very occasionally reported [19], even in according to our knowledge, only one case of OAVS associated with Hirschsprung disease [26] has been reported.
Some patients with OAVS have clinical findings that overlap with other syndromes (i.e. Branchio-oto-renal spectrum disorders, mandibulofacial dysostosis Treacher Collins, Townes–Brocks and CHARGE syndromes) involving structures derived from the first and second pharyngeal arches [19]. Unlike OAVS, individuals with the diagnoses of these type of syndromes typically have symmetric facial malformations. All our cases meet the minimal diagnostic criteria (hemifacial microsomia (asymmetric hypoplasia of facial structures) with preauricular tags or microtia (with or without preauricular skin tags)) and had additional several malformations however none of them had clinical features of the other syndromes as considered in the differential diagnosis.
4. Conclusion
Not all patients with OAVS present with all the common features. These reported four cases supports the hypothesis advancing OAVS as a genetically heterogeneous disorder.
Competing Interests
The authors declare that they have no competing interests.