


1. Introduction
The highly conserved ubiquitin–proteasome system is the principal machinery for extralysosomal protein degradation in eukaryotic cells. The 26S proteasome, a large multi-catalytic multi-subunit protease that processes cell proteins by limited and controlled proteolysis, constitutes the central proteolytic component of the ubiquitin– proteasome system. By processing the cell proteins essential for the development, differentiation, proliferation, cell cycling, apoptosis, gene transcription, signal transduction, senescence, inflammatory and stress response, the 26S proteasome plays a key role in the regulation and maintenance of basic cellular processes. Mutations or changes in these signaling pathways lead to defective transition from G2 to S phase [1]. Given that ubiquitin-proteasome pathway affects multiple cellular process, its inhibition by proteasome inhibitor affects a broader spectrum of proteins with diverse functions. Proteasome inhibitors block protein degradation and cause accumulation of misfolded or damaged proteins, which in turn triggers heat shock response and cell death [2,3]. Furthermore, inhibition of the ubiquitin proteasome pathway (UPP) leads to significant buildup of cytotoxic proteins and activation of apoptotic pathways, particularly in rapidly proliferating cells. Therefore, proteasome inhibitor induces the accumulation of pro-apoptotic proteins in tumorigenic cells but not in the normal tissue [4].
Proteasome inhibitors have shown promising efficacy data in multiple myeloma (MM). Bortezomib, the first-in-class proteasome inhibitor, was subsequently approved by the United States Food and Drug Administration (FDA) as a first-line treatment for newly diagnosed MM patients and for the treatment of relapsed/refractory MM and mantle cell lymphoma[5]. In July 20, 2012, the FDA granted accelerated approval of carfilzomib, a second-generation proteasome inhibitor, for the treatment of relapsed/refractory MM in patients who have received at least 2 prior therapies[6].
It has been reported from non-clinical and clinical trials that proteasome inhibitors inhibit the growth of some solid tumors[3]. The observations from laboratory studies and clinical trials suggest that the anticancer potency for the treatment of solid tumors might be increased if proteasome inhibitor is combined with some conventional anticancer agents [7,8]. Gastric cancer is the second leading cause of death from malignant disease worldwide, with especially high mortality rates in the East, South, and Central Asia; Central and Eastern Europe; and South America [9]. Gastric cancers are most frequently discovered in advanced stages, and the prognosis of advanced gastric cancer remains poor because of limited treatment option. The killing effect of proteasome inhibitor on gastric cancer cells has seldom been studied. The objective of the present study is to explore the effect of proteasome inhibitor Z-Leu-Leu-Phe-CHO (ZLLFC) singly or combined with cisplatin on the proliferation or apoptosis of humnan gastric cancer cell SGC-7901, as well as its influence on the expression level of excision repair cross complementing 1(ERCC1) mRNA.
2. Materials and Methods
2.1 Materials
Proteasome inhibitor Z-Leu-Leu-Phe-CHO was purchased from Sigma Aldrich Chemical Company and dissolved in DMSO as 1mmol/L stock solution. Cisplatin was purchased from QiLu Pharmaceutical Plant. MTT was purchased from Sigma Aldrich Chemical Company. Annexin v-fitc/pi double-staining cell apoptosis kit was obtained from JingMei Bioengineering Limited Company. RTPCR kit was purchased from Fermentas Life Sciences Company. BD FACSCaliburTM System was obtained from BD Company.
2.2 Cell culture
SGC-7901 cells were purchased from Scientific Research institute of Chinese Academy of Science. The cells were cultured in RPMI 1640 medium (Hyclone Fisher Scientific International) supplemented with 10% (v/v) heat inactivated newborn calf serum (Gibco Life Sciences) in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
2.3 Tetrazolium dye methylthiotetrazole (MTT) cytotoxicity assay
SGC-7901 cells were seeded into 96-well microculture plates at 1×103 cells/well(200μl/well) and allowed to adhere for 24h. Then the cells were divided into 6 groups. Group 1 was the control group. The group 2 and 3 were exposed to 0.5μmol/L and 1μmol/L ZLLFC respectively. The group 4 was exposed to varying concentrations of cisplatin(1mg/L, 0.5mg/L, 0.25mg/L, 0.125mg/L, 0.0625mg/L, 0.03125mg/L), and group 5 and 6 were exposed to cisplatin same as the group 4, requiring concentrations of 0.5μmol/L and 1μmol/L ZLLFC added to these two group separately. The cells were then incubated for another 48h or 72h, 20μl of 5mg/ml MTT was added to each well. The plates were incubated at 37°C for 4h and the medium was replaced with 100 ml of DMSO. The absorbance in the control and the drug-treated wells were measured at 490 nm for absorbancy(A value) using microplate reader. Cell viability(%)= [(A value of experimental group-A value of backgound)/(A value of control group-A value of backgound)]×100%. Each experimental data point represents the average value obtained from six replicates, and each experiment was repeated three times.
2.4 Electron microscopy
SCG-7901 cells in log growth phase were digested and adjusted at a cell concentration of 5×106/L, then seeded onto 6-well microculture plates. After allowance to adhere for 24h, the cells were divided into 4 groups including control group, 1μmol/L ZLLFC group, 1mg/L cisplatin group, combined 1μmol/L ZLLFC and 1mg/L cisplatin group. Each group was exposed to the corresponding drug at the above concentrations. After incubated for another 24h, the cells were fixed by glutaraldehyde and osmium tetroxide, dehydrated by ethanol and acetone, and embedded in epoxy resin. Thin sections were stained in uranyl acetate and lead citrate, and examined under transmission electron microscope.
2.5 Quantification of apoptosis
The group division and drug explosure were as the same as that for electron microscopy. The cells were collected, washed in phosphate buffered saline (PBS) and stained by Annex-V and propidium iodide(PI). The samples were then analyzed by fluorescence-activated cell sorting (FACS).
2.6 RT-PCR (reverse transcription PCR) analysis
SCG-7901 cells in the log growth phase were digested and seeded onto 6-well microculture plates. After allowed to adhere for 24h, the cells were divided into 4 groups including the control group, 1mg/L cisplatin group. Each group was exposed to the corresponding drug to the above concentration. After incubated for another 24h, total RNA was extracted. Reverse transcription was performed by reverse transcriptase kit. The resulting cDNA was subjected to the PCR-based amplification with the following oligonucleotides: for ERCC-1, 5’-GGCGACGTAATCCCGACTA-3’ and 5’-AGTCTCCCCAGGCTCTGC-3’; for β-actin, 5’-GTGGG GCGCCCCAGGCACCA-3’ and 5’-CTTCCTTAATGTCACGCAC GATTTC-3’. PCR was performed under the following conditions: 5min 95°C preincubation, followed by 30 cycles of denaturation (45s, 94°C), annealing (45s, 58°C), and extension (1min, 72°C), followed by a final extension of 10 min at 72°C. 5μl PCR products were mixed with 1 μl 6×Loading dye Solution and then electrophoresed with DNA marker, followed by visualization by gel imaging system from Alpha Company. Each experiment was repeated three times.
2.7 Statistic analysis
All the descriptive data was expressed by mean±standard deviation. Statistical analyses were performed using SPSS 11.0. Student’s t-test was used to compare the difference between the two groups and Dunnet-test (ANOVE) was used to compare the difference among three groups respectively. P value of less than 0.05 was considered statistically significant.
3. Results
3.1 Cell viability of SGC-7901 cells treaded by ZLLFC and cisplatin
As shown in Figure 1, cell viability of group 2 or 3 was lower than group 1 (control group)(p<0.01) in a time-dependent manner,but there’s no significant difference between group 2 or 3. As shown in Figure 2 and Figure 3, cisplatin reduced proliferation of SGC- 7901 cells in a dose- and time-dependent manner. Also in Figure 2 and Figure 3, the cell viability of group 5 and 6 indicated that the combined cisplatin and ZLLFC induced more significant inhibitory effect on SGC-7901 cells than that by cisplatin singly, whereas, 0.5 μmol/L or 1μmol/L ZLLFC showed the approximately synergistic action with cisplatin in the inhibitory effect on SGC-7901 cell growth.



3.2 Morphology of SGC-7901 cells treated with ZLLFC and cisplatin
SGC-7901 cells showed normal morphology at normal culture condition by transmission electron microscope (Figure 4a). Treated with 1μmol/L ZLLFC or 1mg/L cisplatin, the cells showed mild apoptotic changes: a few cytoplasmic vacuoles, mild cell shrinkage, increased cell density (Figure 4b, Figure 4c). Combined application of 1μmol/L ZLLFC and 1mg/L cisplatin caused typical apoptosis features on SGC-7901 cells: cell shrinkage, nuclear condensation and chromatin condensation, crescent nucleus, apoptotic bodies aggregated adjacent to the cell membrane, as well as to complete nuclear and cell membrane(Figure 4d).

3.3 Apoptotic rate of SGC-7901 cells induced by ZLLFC and cisplatin
The apoptotic rates of the control group, ZLLFC group, cisplatin group, and combined group were (0.047±0.025)% (4.37±3.00)% (11.78±2.85)% (23.94±2.22)% respectively. The apoptosis rate of the combined group was significantly higher than that of ZLLFC group or cisplatin group (P<0.01) P<0.01)(Figure 5a-Figure 5d).

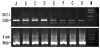
3.4 The ERCC1 mRNA expression level on SGC-7901 cells induced by ZLLFC and cisplatin
The optical density value of ERCC1mRNA in cisplatin group was 717.33±29.17 (Figure 6, lane C,D), much higher than that of the control group(lane A,B, 570.83±20.76)(p<0.01). The optical density values of the two combined groups were 297.33±12.93 (lane E,F, treated with 0.5μmol/L ZLLFC and 1mg/L cisplatin) and 284.83±24.80 (lane G,H, treated with 1μmol/L ZLLFC and 1mg/L cisplatin), which were significantly lower than that of the cisplatin group(p<0.01), but there was no statistical difference between the two combined groups (p>0.05).
4. Discussion
Proteasome inhibitors are a kind of novel anti-tumor agent which can inhibit cell growth and induce apoptosis by inhibiting the function of proteasome system, impacting the degradation of proteins related to cell growth or apoptosis. The first approved proteasome inhibitor, bortezomib belongs to boron-containing compounds, and MG132 is from aldehyde-containing compounds. Bontizomib with its brand name as Velcade has provided promising efficacy data on hematologic malignancies such as MM and mantle cell lymphoma. [5,10,11] MG132 induces apoptosis potentially through the following pathways: formation of reactive oxygen species (ROS), cooperation with APO2L or tumor necrosis factor (TNF)-related apoptosis inducing legand(TRAIL), trimerization of HSF1, NF-KB inhibition, or P53-indepentdent pathway [12,13].
Although bontizomid and carfilzomib have been proved effective in clinical trials and clinical practice, many disadvantages still exist owing to its severe side effects such as decreased efficacy toward solid tumors, interactions with numerous natural products and acquisition of drugresistance in a large portion of cancer patients. The most frequent side effects (incidence >30%) were associated with bortezomib in clinical trials including asthenic conditions such as fatigue, generalized weakness, gastrointestinal symptoms (nausea, vomiting, diarrhea, etc.), hematological toxicity, peripheral neuropathy characterized by decreased sensation, paresthesia and high rate of shingles [12]. In terms of drug-resistance, bortezomib has been reported to have up to 88% response rate, but about 60% of the patients treated with bortezomib develop resistance within an average of one year from the initial treatment [14,15]. Carfilzomib, a “second-generation” proteasome inhibitor, was developed to address bortezomib’s toxicity and drug resistance while achieving comparable potency [16]. These findings encourage basic research to develop newer generation proteasome inhibitors that would broaden the spectrum of activity including as targeting to solid tumor, produce more durable clinical response and more potentiating what to combined with conventional treatment, as well as to provide improved safety profile.
The proteasome inhibitor Z-Leu-Leu-Phe-CHO (ZLLFC) used in our study also belongs to aldehyde-peptides and has similar structure with MG132. Proteasome inhibitors can influence the NF-κb and ERK/ MAPK signal transduction pathway, cell cycle related protein p27kip1 as well as Bcl-2 protein family. Therefore, preteasome inhibitors have the potential to reverse the anti-apoptosis effect initiating by chemotherapeutic agents and become multi-drug resistance(MDR) reversal agents [17,18]. The clinical trials according to the therapeutic function of proteasome inhibitor on solid tumors such as lymphoma, lung cancer, pancreas cancer, gastric cancer,etc. are ongoing.
With MTT cytotoxicity assay, electron microscopy and quantification of apoptosis, our study showed that proteasome inhibitor ZLLFC used singly could inhibit the growth of gastric cancer cell SGC-7901, induce them into early apoptosis, while, there’s no significant difference in promoting apoptosis effect between 0.5μmol/L and 1μmol/L ZLLFC(p>0.05). MTT cytotoxicity assay indicated cisplatin could reduce proliferation of SGC-7901 cells in a dose-dependent manner. Combined ZLLFC and cisplatin of different concentration gradient showed lower cells viability than cisplatin used singly. Meanwhile, the growth inhibitory effect in the combined group have a time-dependent manner, ie, drug action of 62 hours showed stronger effect than that of 48 hours. With 0.5μmol/L or 1μmol/L ZLLFC, the combined group showed inhibitory effect to the same extent. These results suggested the cytotoxic effect of ZLLFC on SGC-7901 cells. When combined with cisplatin, ZLLFC increased the cytotoxicity and promoting apoptosis effect of cisplatin in a time-dependent manner ,to achieve the best concentration would needd more investigation.
Cisplatin is a wildly used anti-tumor agent, it damages DNA and inhibits DNA replication by cisplatin-DNA adduct. Cisplatin used singly is liable to induce drug resistance,one of the major mechanisms of drug resisitance induced by cisplatin is injured DNA-strand incision [19]. ERCC1(excision repair cross complementing 1) is a NER(nucleotide excision repair) component that participates in DNA damage recognition and DNA strand incision. ERCC1 plays an importantrole in the repair of damaged DNA and maintaining the integeity of genetic information.The inhibition of ERCC1 expression by antisense approaches has been reported to reduce cisplatin-DNA adduct repair [20]. In human ovarian and gastric cancer, elevated ERCC1 mRNA expression in the tumor tissue has been related to increase cisplatin resistance [21,22]. Overexpression of ERCC1 has also been associated with resistance to cisplatin in human hepatocellular carcinoma [23]. Li Y, et al. treated gastric cancer cells withlactacystin and Mimnaugh EG, et al. treated ovarian carcinoma cells with combination of cisplatin and proteasome inhibitor lactacystin(LC) or N-acetyl-leucyl-leucyl-norleucinal (ALLnL),both proteasome inhibitors prevented the increase in ERCC1 mRNA expression that occured in cells exposed to cisplatin [24,25]. Low ERCC1 is candidate for predictive biomarkers for first-line in advanced gastric cancer [26] and patients who suffer from gastric cancer with ERCC1-negative expression could benefit more from adjuvant chemotherapy [27]. Our study also indicated an increase of ERCC1 mRNA expression in SGC-7901 cells when exposed to cisplatin. But combined ZLLFC and cisplatin significantly decreased the ERCC1 mRNA expression in comparison with cisplatin used singly (p<0.01). This result suggested ZLLFC could prevent the up-regulation of ERCC1 transcription level and might be a good treatment in gastric cancer.
The discovery of less toxic and novel proteasome inhibitors may provide more viable drug candidates for the treatment of cancer patients in the future. Our study indicated a novel drug candidate of proteasome inhibitor, Z-Leu-Leu-Leu-CHO(ZLLFC), could induce SGC-7901 cells apoptosis. ZLLFC reversed the drug resistance of cisplatin by down-regulating ERCC1 transcription level, which is the key point of cisplatin-DNA adduct incision. Throughout the study, there was no significant difference between 0.5μmol/L and 1μmol/L ZLLFC. More research in vitro studies with different tumor cell lines and animal models should be warranted to offer adequate basic research data to pursue potential clinical development for this asset in the future.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
Thanks to Dr. Zhang QH (Shanghai Biochip Co., Ltd.) and his team for their expert technical assistance. We are also grateful to Dr.Wu XN to revise this article. The English version in this document has been checked by at least two professional editors, both native speakers of English,we give our thankfulness to them.