



1. Introduction
Hemoglobin and albumin are major circulating proteins in the blood and exert determinant biological functions. The structures and functions of hemoglobin and albumin can be altered by oxidative stress which role in pathology progression is now being well established [1]. Non-enzymatic glycation of these proteins constitutes one of the underlying modification factors that occur in diabetes mellitus. It is the result of covalent binding of glucose to amino groups of such circulating proteins and also proteins from the extracellular matrix [2].
The glycated hemoglobin (HbA1c) level is considered as one of the main clinical parameters used for monitoring long term (2-3 months) glycemic exposition in the blood [3]. With a shorter halflife than hemoglobin in blood, glycated human serum albumin (HSA) appears to be an alternative marker for glycemic control as it can indicate blood glucose status over a short period (2-4 weeks) [4, 5]. Glycated albumin can notably constitute a better glycemic control state in several diabetes-associated pathologies compared to glycated hemoglobin, because it is not affected by abnormal haemoglobin metabolism encountered in some case of type 2 diabetes [6-8]. In addition, through the IMA (ischemia-modified albumin) index evaluation, albumin appears to be a relevant biochemical marker in the diagnosis of numerous complications including myocardial ischemia and skeletal muscle damage [9,10].
Human serum albumin (HSA), as the most prominent protein in plasma, is the major carrier of various metabolites in blood throughout the body, such as fatty acid and also therapeutic drugs. This protein represents also a very abundant and important circulating antioxidants which contribute to maintain the extracellular redox homeostasis [11]. In vivo, the proportion of glycated albumin in healthy people is in 1% to 10% range [12,13] and in the case of diabetes mellitus; this proportion can increase two- to three-fold [14]. Elevated levels of glycated albumin in cases of diabetes mellitus can lead to irreversible damage associated with metabolic disorders such as retinopathy, nephropathy, neuropathy and coronary artery disease [15,16]. Numerous studies have reported the impact of diabetes situation in alteration of many intrinsic biochemical and functional properties (redox state, antioxidant, binding and esterase properties) of human albumin in vitro and in vivo models, as well [17-19]. These studies have notably suggested the possible clinical relevancy of albumin as a new biomarker of diabetes and associated pathologies. Many other studies have also pointed out that bound fatty acid could interfere on several albumin properties previously mentioned such as drug binding [20 , 21]. It is known that some fatty acids can have direct competition with drugs or lead to allosteric effects during the binding of drugs and other solutes with albumin [22-24]. The determination of specific functionality of albumin such as affinity for drugs of steroid hormones could require a preliminary step of delipidation.
Therefore, the aim of this study was to find a reliable and convenient method for albumin delipidation and purification from plasma which does not significantly impact the redox state, intrinsic structural and functional integrities of the protein. This method of albumin isolation and delipidation needs to be time-efficient and technically less demanding in order to be suitable for clinical practice.
2. Materials and Methods
2.1 Chemicals and reagents
Human serum albumin 99% (A1653), human serum albumin fatty acid free (A1887), bicinchoninic acid (B9643), nitroblue tetrazolium (NBT) (N6639), 2,4-dinitrophenyl hydrazine (DNPH) (D2630), p-nitrophenyl acetate (PNPA), ketoprofen and 8-Anilino- 1-naphthalenesulfonic acid (ANSA) were all purchased from Sigma- Aldrich (St Louis, MO). Serum samples from non-diabetic subjects (% HbA1c = 5.5 ± 0.3, N=10) were obtained by the Biochemistry laboratory of the Centre Hospitalier Universitaire (CHU, Saint-Denis, La Reunion).
2.2 Purification of HSA from plasma by affinity chromatography
Blue Sepharose 6 Fast Flow was used to isolate HSA from serum of patients. The gel was packed into a column (4.3 × 0.8 cm), washed with starting buffer (Tris HCl 50 mM, pH 7.2), and equilibrated with 10 bed volumes of starting buffer. The gel was then transferred into tubes (2.0 mL) or packed into a column (10 mL). The different steps of the chromatography (binding, washing and elution) were achieved by column chromatography (Col Chrom) and also by the technique of batch separation (Batch Chrom). The protocol of column chromatography was well described in a previous study [19]. For the chromatography by batch treatment, plasma (0.5 mL) was applied to the gel (1 mL) and left with weak stirring for 1 hour at 4°C. After the first centrifugation (4000 g 10 minutes), the supernatant was discarded. The gel was washed six times with 1 mL of the starting buffer, in the same conditions. The gel-bound albumin was then eluted six times with 1 ml of 50 mM Tris-HCl buffer (pH 7.4) containing 1.5 MM NaCl. All HSA fractions were mixed and were washed with 0.1 M sodium phosphate buffer (pH 7.4) in order to remove NaCl and finally concentrated by ultrafiltration (Amicon 10K, Millipore).
2.3 Purification of HSA from plasma by AS precipitation
Human albumin was also purified from serum by precipitation with ammonium sulphate (AS) using a two-step protocol [28]. In the first step, saturated ammonium sulphate solution (767 g/L, pH 7.4 adjusted with NH4OH) was added to the serum sample while gently stirring until a concentration of 54% AS was reached. The precipitated globulins were then removed after centrifugation (5000g, 10 min) and the supernatant was collected. The second step consists of an addition of saturated ammonium sulphate solution in the supernatant until a final concentration of 70% AS was reached. The precipitated HSA was collected after centrifugation (10000g for 30 min) and supernatant removed. The HSA pellet was suspended in 0.1 M sodium phosphate (pH 7.4). HSA solution was washed three times to remove AS trace and was finally concentrated by ultrafiltration (Amicon 10K, Millipore).
2.4 Delipidation of HSA
The ethanol–ether extraction method was used to perform the complete delipidation of plasmatic purified albumin [29]:
Firstly about 6 ml of HSA sample (30 g/L) was prewashed with 5% aqueous methanol through Amicon ultrafiltration units (Amicon 10K, Millipore) in order to remove salts and other low molecular mass contaminants. The following steps were performed on albumin aliquots (250 μL) : each aliquot was mixed with 500 μL ice cold ethanol: diethyl ether 3:1 (v/v), incubated at -25°C (2 h) and centrifuged at 4°C and 6000 rpm (30 s). The supernatant (which contained the lipids) was discarded and the remaining protein pellet was suspended with 500 μL ice cold ethanol: diethyl ether 3:2 (v/v) and incubated at -25°C (1 h) before a centrifugation at 10,000 rpm (5 min). Then the remaining protein pellet was suspended in PBS buffer.
After albumin purification or delipidation process, protein and albumin concentrations were determined by using bincinchoninic acid (BCA) and bromocresol green (BCG) reagents, respectively.
2.5 Biochemical characterization of HSA
The 2,4,6-trinitrobenzenesulfonic acid (TNBS) assay is a sensitive method for determining the primary free amino groups in proteins [30]. This method was described in detail in a previous study by our group [31].
Thiol groups level in HSA samples was measured by Ellman's assay using 5, 5' -dithiobis, 2-nitrobenzoic acid (DTNB) [32] as described previously [33]. A standard curve was performed using various concentrations of L-cysteine (10 to 100 nmol) (Sigma). The content of thiol groups was measured in triplicate by reading the absorbance at 412 nm. Results are expressed as the number of free \SH groups per mol of HSA.
Carbonylated level was performed on HSA samples as described before [34]. Level of carbonylation was determined by spectrophotometric assay based on recognition of protein bound DNPH in carbonylated proteins with an anti-DNP antibody [35]. About 200 μg of proteins were successively precipitated in 10% TCA solution, suspended and incubated for 10 min with DNPH solution (0.2% in 2 M HCl). After another precipitation in 10% TCA followed by a centrifugation and successive washes with ethanol–ethyl acetate solution (50:50), the dried pellets were dissolved in a 200 μL guanidine solution (6 M in 500 mM KCl pH 2.5). The absorbance of protein samples was measured at 276 nm and 370 nm for protein and carbonyl contents, respectively. Carbonyl was expressed as mol carbonyl/mol HSA.
2.6 Structural characterization of HSA
Fluorescence spectra were carried out on a FluoroMax®-4 (Horiba) spectrophotometer with protein samples at a concentration of 10 μM in PBS. The tryptophan emission spectra were obtained in the range of 250-600 nm under excitation at 270 nm. All fluorescence spectra were normalized to protein concentration with the respective different absorption spectra. 1-anilino-8-naphthalenesulfonic acid (ANSA) was dissolved in 5 μM albumin samples with an ANSA/albumin ratio of approximately 0.4 (mol/mol) in order to be sure that the dye was linked to the hydrophobic sites of albumin. ANSA-protein complexes emission fluorescence spectra were performed in the range of 350-700 nm under an excitation wavelength of 470 nm.
Migration profiles of purified albumin and plasma were analyzed by SDS- Polyacrylamide gels (12% of acrylamide) and stained by Coomassie blue according to Laemmli’s method [36].
2.7 Red blood cells hemolysis test
The antioxidant properties of modified albumin preparations were analyzed with the free radical-induced blood hemolysis test [37]. Human blood samples were obtained from the Biochemistry laboratory of the Centre Hospitalier Universitaire (CHU, Saint-Denis, La Réunion) and were taken on EDTA substrate as anticoagulant. Then, plasma was removed and erythrocytes were washed with an isotonic solution (NaCl 0.15 M). Each well of a 96 well-plate was filled with 100 μl (about 1.108 erythrocytes, 400 000 cells/μL final concentration) of diluted solution of red blood (1/10 in 0.15 M NaCl). Different albumin samples (10 μM final concentration) were added in triplicates. Hemolyses were started by adding 0.45 M of AAPH in each well. Turbidimetry at 450 nm was recorded every 10 min using a 37°C-thermostated microplate reader. Results were expressed as 50% of maximal hemolysis time (HT50 in min) which represents the total defense against free radicals in human and animal models submitted to oxidative stress [38].
2.8 ORAC test
The oxygen radical absorbance capacity (ORAC) developed by Ou et al. [39] assay has been widely accepted as a standard tool to measure the antioxidant activity in the nutraceutical, pharmaceutical, and food industries [40]. The ORAC assay using fluorescein as the fluorescent probe measures the antioxidant capacity of samples to protect from oxidative damage of fluorescein initiated by AAPH. The automated ORAC assay was carried out on an Infinite M200 pro spectrofluorometric analyzer (TECAN) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The reaction was carried out in phosphate buffer (75 mM, pH 7.4) containing each albumin sample (25 μL) in six replicates and fluorescein solution (80 nM, 150 μl). Trolox solutions at different concentration (6-50 μM) were used as calibration standards. The fluorescence kinetic of fluorescein was followed for 1 hour at 37°C after adding AAPH solution (150 mM, 25 μL) in each well. The fluorescence was recorded every 2 minutes. Antioxidant curves (fluorescence vs. time) were first normalized to the curve of the blank (phosphate buffer instead of samples or trolox). Then the area under the fluorescence decay curve (AUC) was determined. The antioxidant activity (ORAC value) of samples was calculated by using a trolox calibration standard as follow:
2.9 Albumin affinity for ketoprofen: experimental design of the fluorescence spectroscopy method
This method is based on the quenching of albumin fluorescence induced by its interaction with drugs [41]. The intrinsic fluorescence of human albumin is mainly attributed to the tryptophan residue (Trp-214). Different series of assay solutions were prepared by mixing 20 nmol of different HSA preparations with ketoprofen at variable concentrations ranging from 0 to 160 nmol. Each solution was heated for 30 min at 37°C and transferred into a quartz cell. The fluorescence spectra were recorded in the range of 250-500 nm under excitation at 270 nm. The binding parameters (i.e. the binding constant KA and the binding site number (n) for ketoprofen were obtained from the equation given below:
Where F0 and FC are the tryptophan fluorescence intensities in the absence and presence of a drug at concentration [C], respectively, and KA is the constant formation of the complex formed between the drug and albumin, expressed as L/mol.
2.10 Experimental design of esterase-like activity
The reaction of p-nitrophenyl acetate with HSA was followed spectrophotometrically at 400 nm with an Infinite M200 pro spectrofluorometric analyzer (TECAN) by monitoring the absorbance of p-nitrophenol. The reaction mixtures contained 5 μM p-nitrophenyl acetate and 20 μM HSA in 67 mM sodium phosphate buffer (pH 7.4). Reactions were followed at 25°C. Under these conditions, pseudofirst- order rate constant analysis could be applied, as described in previous reports [42,43], and an apparent hydrolysis rate constant (kobs) was calculated [44].
2.11 Statistical analysis
The data are expressed as the means ± standard deviation (SD) from a minimum of three experiments. Statistical significances were determined using one-way ANOVA (followed by the Student’s t test) for multiple comparisons; a p value of less than 0.05 was required for significance.
3. Results
In the first stages of the study, human albumin was isolated from a pool of plasma by using two different techniques: the first technique used a single-step affinity chromatographic method based on the specific interactions between HSA and a ligand. Here, the textiles dye Cibacron Blue F3G-A was used as ligand. The second procedure, using ammonium sulphate (AS) is based on the difference in solubility of HSA and other plasmatic proteins.
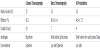
Table 1 gives a characterization of the three techniques of albumin isolation (column chromatography, batch chromatography and AS precipitation) in terms of efficiency, timeframe, advantages and inconveniences. If the column chromatography technique gives the best yield compared to other protocols (68.1 %), this procedure appears to be time and plasma- consuming. The isolation of albumin by batch chromatography and AS precipitation methods could be realized more rapidly (few hours) and with a very low quantity of plasma (about 0.5 mL) but gives a lower yield (about 45%) which could be improved. As shown in Figure 1a, the native electrophoretic pattern did not give convincing differences between main fractions of purified HSA isolated by the different protocols.However, a band related to the dimeric form albumin (around 132 kDa) was found to be more expressed for HSA fractions isolated by AS precipitation and also by column chromatography.

Following the isolation step from plasma, HSA fractions sourced from the three methods of purification were characterized as featured in Figures 1(b-d). With average values of 25.0 moles of free primary amine and 0.35 mole of free thiol (per mol of HSA), all albumin fractions exhibited the same biochemical pattern. Similarly, all HSA fractions displayed a quite similar spectral pattern of the tryptophan fluorescence emission with a slight blue shift (around 6-7 nm) of the maximum of fluorescence in comparison with commercial HSA spectra.
After these first steps of purification, HSA isolated from plasma with the AS precipitation technique was defatted. This delipidation method which needs to be inexpensive and reproducible combines several steps of precipitation with ethanol: diethyl ether and centrifugation. Biochemical, structural and functional properties concerning fatted HSA (FAHSA) and defatted HSA (D-HSA) were investigated and compared with commercial human serum albumin (with and without fatty acids).
Figure 2 features the primary amine, free thiol and carbonyl levels for commercial (FA-HSAC and D-HSAC) and purified albumins (FAHSA P and D-HSAP). With average values of free primary amine around 25.0 moles for HSAP and 20.0 moles HSAC, delipidation process did not affect lysine and arginine residues (Figure 2a). In addition, all albumin samples exhibit a quite similar redox state with carbonyl levels around 0.45 mol (/mol HSA) (Figure 2b). To summarize, these biochemical parameters did not significantly differ between fatted and defatted albumins. If free thiol levels remain around 0.35 mole (per mol of HSA) for FA-HSAC ,D-HSAC and FA-HSAP, this level was significantly higher and above 3 mol for defatted HSA (D-HSAP) (Figure 2c).

In Figures 3, the normalized spectra for the tryptophan and 1-anilino-8- naphthalenesulfonic acid (ANSA) fluorescence are reported. The typical bands observed in Figure 3a, at about 340 nm is attributed to the sole albumin tryptophan (Trp-214). The absence of fatty acid in HSA (D-HSAC and D-HSAP) is associated with an increase of this fluorescence emission and a very slight red shift (around 1-2 nm) of the maximum of fluorescence compared with non-defatted albumins. The conformation changes in HSA hydrophobic regions were detected with the fluorescent probe ANSA (Figure 3b). If no difference in ANSA fluorescence emission was noticed between fatted and defatted forms of purified HSA (HSAP), delipidation of commercial HSA (HSAC) induced a decrease in its extrinsic fluorescence. In addition, a marked red shift (7-8 nm) of the maximum of fluorescence was noticed after delipidation of commercial HSA, whereas this shift was less pronounced between FA-HSAP and D-HSAP (around 3 nm).

Three major intrinsic properties of albumin, which are highly dependent on biochemical and structural integrity of the protein, were also further investigated: antioxidant, affinity and esterase properties. The antioxidant properties of commercial and purified albumin were first investigated by using two free radicals scavenging capacity assays. The red blood cells hemolysis test is based on the intrinsic capacity of albumin to protect erythrocytes from free radical-induced hemolysis. The results reported in Figure 4a showed a higher and a similar hemolysis half-time (HT50) for all albumin samples (around + 60%, p<0.01 vs. PBS control) reflecting the antioxidant property exerted by the plasmatic protein. The second antioxidant test named ORAC, showed an increase of ORAC values for both forms of defatted HSA (D-HSAC andD-HSAP) which did not reach statistical significance (Figure 4b). This last test strengthens the previous results concerning quite similar antioxidant properties for all HSA samples. These both results attest the protected action of albumin is not impacted by the delipidation (for HSAP) or the absence of fatty acids (for HSAC).
The impact of delipidation of HSA on the drug binding and esterase activities were subsequently investigated. Ketoprofen constitutes a site-selective probe of Suddlow site II and its interaction with albumin was evaluated using a method based of tryptophan fluorescence emission quenching. This method allowed to determine the binding constant (Ka) between albumin and its ligand. In figure 5a, are reported Ka values for all HSA samples. A marked and significant increase in affinity was noticed for defatted purified albumin (D-HSAP) (Ka= 4.27.109 L/mol) in comparison with albumin control (FA-HSAP) (Ka= 0.49*109 L/mol). If an enhancement of this affinity was also observed for commercial albumin fatty acids free (DHSAC), this increase did not reach statistical significance.
Similarly to the drug binding capacity, esterase-like activity of albumin plays an important role in the pharmacological properties of therapeutic drugs. Esterase activities of HSA samples were investigated by monitoring the hydrolytic conversion of p-nitrophenyl acetate to p-nitrophenyl. The kobs values reflecting this activity are featured in Figure 5b. Both forms of purified HSA (FA-HSAP and D-HSAP) exhibited quite similar kobs values around 5.0*10-5 s-1, while this esterase activity appears to be higher for both commercial albumin ( kobs= 1.20*10-4 s-1 for FA-HSAC and kobs= 1.45*10-4 s-1 for FA-HSAC).
4. Discussion
Because of its high abundance in extracellular compartment, albumin is considered as the major metabolite carrier and antioxidant in the plasma and plays a key role in plasmatic homeostasis. The plasmatic compartment is highly exposed to continuous and deleterious phenomenon (oxidative stress, glycation, carbamylation…) and its biochemical characterization could represent a fingerprint of the physiological or pathological situation of a patient. The discovery of new biomarkers constitutes a real challenge in detection, prediction and the monitoring of diabetes risk and associated metabolic disorders progression. Numerous clinical studies pointed out the relevancy of glycated albumin as a suitable marker of glycemic control in numerous physiopathological states such as hemodialysis patients [4], gastrectomized subjects [45] and gestational diabetes [46]. Similarly, ischemia-modified albumin (IMA) is considered as a promising biomarker for the evaluation of ischemic events [47], but also for cerebrovascular diseases [48], pulmonary embolism [49] and liver cirrhosis [50].
All these studies illustrate the relevance of albumin as a key biochemical parameter in monitoring and evaluation diabetes related metabolic disorders. The biochemical, structural and functional characterisations of plasmatic albumin still warrant further investigations in clinical studies. To gain good insight in albumin modification analyses in pathological situations, a strategy for albumin purification needs to be engaged. The defined protocol should be efficient, rapid and should lead to a non-altered albumin in terms of structural and functional properties.
In our experimental conditions, we showed that the three selected isolation methods did not differ in terms of biochemical parameters for purified albumin. Isolated albumins displayed about 24-25 free amine groups which is a quite similar level observed for commercial HSA obtained by heat-shocked fractionation [19]. The redox state of the protein, probed by the Ellman’s assay was not drastically impacted regardless the type of purification and in comparison with commercial HSA. With a 0.40 mol value, the levels of free sulphydryl group of purified albumins are however found to be lower than expected in HSA from healthy subject (about 0.7) [51]. In addition, numerous studies have reported a significant oxidation of thiols in commercial albumins which could drop to 0.15 mol [19,33]. This comparison suggests that our purification protocols have a minor deleterious impact for the biochemical albumin integrity than the above mentioned studies. The tertiary conformation analysis monitored through the tryptophan fluorescence emission was in perfect agreement with this remark. No difference in the spectral pattern was evidenced between our purified HSA preparations, reflecting a similar structural conformation. Conversely, the relative blue shift observed in comparison with commercial HSA fluorescence spectra could be explained by a less polar environment of the tryptophan attesting that the local tertiary conformations of the purified proteins differ from the commercial one. Consequently, these conformational changes have a direct impact on some biological function of albumin. Recently, affinity capacities and esterase activities have been found to be markedly altered in albumin isolated from plasma by batch chromatography method in comparison with commercial HSA (unpublished data). This impairment of esterase activity of purified albumin is confirmed in the present study.
As observed on native PAGE electrophoresis, the presence of dimmers and even oligomers in purified albumin highlight the heterogeneity of the plasmatic protein isolated by different techniques. It is well known that typical albumin samples contain a portion of dimeric form [52,53]. The proportions of these dimers could be variable (between 1.4 to 15.4 %) and are very depending on the protein purification procedure [12].
In a second part of the study, it was of utmost importance to avoid the presence of fatty acids in albumin molecules which could diminish its affinity for other metabolites 19 including therapeutic drugs, steroids and bilirubin [12]. For illustration, fatty acids could notably influence the affinity of serum albumin for cobalt in patients with fatty liver and could consequently interfere in the IMA measurement [54]. In clinical investigation, albumin samples could be hindered by the lipid content which can vary strongly between patients. Consequently, it was crucial to develop a reproducible processing procedure for albumin delipidation with no impact on albumin properties. In our experimental conditions, ethanolether solution was used for the delipidation process. Taken together, main results showed that this process have clearly a deleterious impact on biochemical, structural and functional properties of albumin because of the use of organic solvents. We can hypothesise that the affinity increase of defatted HSA for ketoprofen could be attributed to an enhanced interaction of the protein with the drug in the absence of fatty acids. But, the indisputable change in the tertiary conformation of albumin favoured by a possible disulfide bond disruption, induced by this process, could also explain this renewed affinity. Surprisingly, this delipidation process did not have any impact on intrinsic antioxidant properties which are mainly imputable to the scavenging activity of Cys-34. It is clear that this residue was not affected by the albumin delipidation which did not induce any oxidative modification. Similarly, the change in albumin conformation particularly around its hydrophobic pockets did not induce any significant impairment of the esterase activity which however involves some residues located in hydrophobic site II of Suddlow [44]. These residues such as Arg-411 or Tyr-410 seemed to be protected against the deleterious effects of delipidation process.
If fatty acids removal by delipidation did not impact the esterase like activities of HSA, significant differences in enzymatic activity were clearly noticed between commercial or purified albumins. Indeed, the esterase activities were found to be drastically impaired in both forms of purified HSA in comparison with the corresponding commercial albumins. If the protocol for albumin purification did not significantly impact main biochemical, structural and functional parameters (antioxidant and affinity properties), certain activities such as esterase appeared to be more sensitive.
5. Conclusion
In this study we have shown that the two-step precipitation of HSA with ammonium sulphate (AS method) enabled to purify albumin having preserved structural and functional properties and through a rapid and efficient protocol. The requirement for the analysis and discovery of new albumin related biomarker in a large panel of patients stresses the importance of developing a simple, reliable and technically less demanding method of HSA isolation. The method proposed here appears to be suitable for clinical practice. However, the delipidation step following the purification should not to be considered.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
All the authors substantially contributed to the study conception and design as well as the acquisition and interpretation of the data and drafting the manuscript.