


1. Introduction
Rheumatoid Arthritis (RA) is an chronic systemic autoimmune disease that affects approximately 1% of the adult U.S. population [1]. The pathophysiology of RA involves chronic inflammation leading to proliferation of the synovium with subsequent cartilage damage and bony erosions [1]. Ocular inflammation is an extra-articular manifestation of RA and can be the initial presentation of RA [2]. It is important for the rheumatologist to understand the ocular diseases associated with RA such as keratoconjunctivitis sicca (KCS), episcleritis, scleritis, peripheral ulcerative keratitis (PUK), anterior uveitis, as well as retinal vasculitis [3]. In fact, any component of the eye can be affected in RA. However, anterior eye involvement is the most frequently encountered in clinical practice [4].
2. Keratoconjunctivitis Sicca
Keratoconjunctivitis sicca (KCS) or Dry Eye Disease (DED) is an ocular condition of inadequate lubrication of the eye caused by decreased tear production and/or excessive fluid loss from the eye surface in the presence of normal tear secretion, producing irritation of the eye accompanied by symptoms of soreness, grittiness and dryness [5]. In RA, DED is most likely due to aqueous deficient state of the ocular surface however, evaporative DED can coexist as a continuum [6].
2.1 Epidemiology
KCS is the most commonly associated ocular manifestation of RA and is often the initial manifestation of ocular disease [7]. The reported incidence and prevalence of DED in patients with RA ranges from 18% to 90% with moderate to severe symptoms seen in up to 50% of patients [3,8]. The variation in the reported rates of KCS can be attributed to factors such as: limited sample size, age of patients sampled, gender, subjective versus objective determination of DED. The association between RA and KCS is well established and contributes to morbidity [4-6].
2.2 Features and pathogenesis
Symptoms of KCS include: burning, gritty sensation and dryness. A recent meta-analysis demonstrated a link between dry eye disease and depression and anxiety [7]. If left untreated, KCS may lead to a lidwiper epitheliopathy (analogous to that found in long-term contact lens wearer), keratitis, and possibly even opacification of the cornea [8]. In severe cases, one can see paracentral corneal melts which can cause permanent visual loss, and even perforation.
Inflammation has been postulated as the underlying cause for KCS [9]. The pathogenesis of RA involves an inappropriate attack on the synovial membrane by monocytes, which mature into macrophages and secrete cytokines such as interleukin-1 (IL-1) and tumor-necrosis factor (TNF). These cytokines, in addition to recruiting other cells to attack the joints, also begin a cascade of systemic events, which may be the link between RA and KCS. Though the exact mechanism is largely unknown, there is evidence that oxidative stress created by inflammation may lead to destruction of the ocular surface epithelium and/or lacrimal tissue with functional decline in tear production [10,11]. Studies suggest that the oxidative stress on the ocular surface may be the underlying cause for the damage of the lacrimal tissue and subsequently the decline in tear production in patients with inflammatory conditions such RA [10,11]. Moreover, the tears of healthy controls compared to DED patients differ in the inflammatory and anti-inflammatory cytokine balance [12,13]. The tears from healthy individuals have anti-inflammatory components which help maintain the immune-privileged status of the eye [12]. Persistent tear hyperosmolarity favors increased concentrations of inflammatory cytokines, such as TNF-α, IL-1β and IFN-γ and matrix metalloproteinases (MMPs) [14]. Innate immunity also plays an important role as neutrophilic infiltration and neutrophilic DNA extracellular traps have been detected on DED with by-products of neutrophil activation possibly contributing to DED [15]. Corneal damage by alkali burn leads to increased production of inflammatory mediators such as IL-1β, IL-6, MMP and CXCL1 (C-X-C Motif Chemokine Ligand 1), a neutrophil activating protein; and corneal opacity is associated with neutrophil infiltration likely mediated by CXCL1 [16]. Macrophages, in particular M-1 by affecting inducible NO Synthase and IL-18 levels and Natural Killer cells, via CD4Th17 modulation have also been demonstrated to play a role in the pathogenesis of DED [17,18]. Recruitment of CD4 and CD8 T cells have been demonstrated in patients with autoimmune and non-autoimmune DED with increased expression of Intercellular Adhesion Molecule 1 (ICAM) in the conjunctival epithelial cells [19].
Strong expression of IL-23/Th17 axis and related cytokines such as Il-6, IL-23, TGF-β2, and Retineic-acid-receptor-related orphan nuclear receptor γ (RORγt), one of the master regulators in the development of T helper 17 cells (Th17 cells) have also been shown to play a role in DED [20-23].
2.3 Diagnosis
Early diagnosis of KCS is extremely important as therapy may begin at a point when the natural course of the disease may be slowed and devastating corneal complications may be avoided [24]. Unfortunately, there is no single “gold standard” diagnostic test for dry eyes. The diagnosis is often made by obtaining the history and physical exam. Questionnaires are commonly used to guide the history followed by slit lamp exam and fluorescein staining, and Schirmer test [25]. All these exams have drawbacks; therefore, a combination of studies is often recommended to attain a comprehensive evaluation. Recently, more advanced tests have been developed to assess tear osmolarity and tear film stability. These tests are likely to become the standard for assessing dry eye disease in the future.
Subjective tests such as questionnaires are economical and easy, but they inherently lack consistency as patient responses may vary. Moreover, objective severity of the dry eyes cannot be assessed and patients in the early stages of dry eye disease are missed [26]. Fluorescein staining is more likely to diagnose KCS at an early stage, as it is a sensitive indicator of epithelial integrity. Fluorescein is a water-soluble substance that can traverse through damaged corneal epithelial cells, accumulating within the intracellular spaces. In KCS there is an accumulation of fluorescein in the intrapalpebral conjunctiva and cornea likely due to epithelial tight junction dysfunction and cell death [27]. The major drawback to fluorescein staining in the diagnosis of KCS is the lack of specificity as staining of the cornea may also occur in normal eyes. The Schirmer test involves placing a small strip of paper bent at 90 degrees into the lower fornix of the eye and leaving it there for either 2 minutes or 5 minutes [28]. In a normal test the average secretion of tears should be greater than ≈12 mm after 2 minutes, and ≈ 16mm after 5 minutes [28]. However, the Schirmer test has been shown to have sensitivity as low as 25% [29].
A common feature of dry eyes is an increase in osmolarity. Investigations using tear osmolarity have shown a sensitivity of 73% and specificity of 92% in detecting dry eyes [25]. Values greater than 308 mOsms/l are indicative of early/mild dry eye disease, and values greater than 312 mOsms/l indicate advanced/severe forms of dry eye disease. Because the window between mild and severe disease is so narrow (a difference of only 4 mOsms/l) it is imperative that the test is performed precisely. Assessment of tear stability is an alternative means of diagnosing dry eye disease. The most commonly used method is the tear break-up time, but non-invasive tear break-up time tests have also been produced using interferometry, aberrometry, corneal topography, confocal microscopy, and functional visual acuity assessment [26]. Although there is no standard for the diagnosis of dry eye disease, a combination of the above-mentioned tests is more sensitive and specific for the diagnosis of dry eyes.
2.4 Management
Once the diagnosis of KCS is made, the first step includes patient education, modification of the local environment and if possible, a diet richer in essential fatty acids, followed by non-preserved ocular lubrication with artificial tears [30,31]. Referral to ophthalmology will facilitate further interventions such as tear conservation maneuvers via punctal occlusion, overnight ointments, moisture chamber goggles, and prescription drugs containing topical steroids and secretagogues [31]. However, since inflammation is the driving force behind KCS, we must provide anti-inflammatory therapy to alter the disease course. Some of the anti-inflammatory drugs that have been studied in KCS include cyclosporine A (CsA), corticosteroids, disease modifying anti-rheumatic drugs (DMARDs), and biologics.
2.5 Clinical trials impacting management of KCS
Sixty-nine clinical trials have been conducted to evaluate cyclosporin, an immunomodulator with anti-inflammatory effects, in keratoconjunctivitis sicca. In 2002, the FDA approved ophthalmic CsA 0.05% (Restasis®) for KCS, based on the results from four multicenter, randomized, clinical studies performed in about 1,200 patients with moderate to severe KCS. CsA at 0.05% demonstrated statistically significant increases in Schirmer testing for tear production at six months in 15% of the subjects treated versus compared to 5% of vehicle treated patients. No increase in tear production was observed in subjects being treated with anti-inflammatory drugs or using punctal plugs. However, in clinical practice CsA at 0.05% (Restasis®) has proven to be only minimally effective [32,33].
In 2016, (Xiidra®) lifitegrast was approved for the treatment of KCS. Lifitegrast is an active inhibitor of both leukocyte functionassociated antigen-1 (LFA-1) and the intracellular adhesion molecule- 1(ICAM-1), both of which mediate migration and adhesion of the white blood cells to sites of inflammation.
In 2018, the FDA approved cyclosporine A ophthalmic solution 0.09% (Cequa®), a patented, novel, preservative-free nanomicellar formulation. This new ophthalmic aqueous formulation provides a high concentration of CsA while allowing better solubility and penetration of the eye's aqueous layer. During the phase 2b/3 of the clinical trial, 455 patients were studied demonstrating increased tear production and good tolerance. During phase 3 of the trials, 744 patients with DES were treated with Cequa® or its vehicle. After 12 weeks of treatment, the patients on the nanomicellar CsA showed statistically significant improvement in tear production as measured by Schirmer's score compared with vehicle [31,35].
Improvements in ocular staining assessments were also seen as early as one month after. Common adverse effects were instillation site pain (22%) and conjunctival hyperemia (6%) with < 5% of the patients reporting eye irritation, blepharitis or headache.
Other on-going clinical trials include:
The use of loteprednol etabonate (LE) before the initiation of topical cyclosporine A (tCsA) therapy in patients with mild-to-moderate dry eye disease was evaluated in a prospective, multicenter randomized double-masked clinical study. 118 patients were randomized to receive either LE and tCsA (n=61) or artificial tears (AT) and tCsA (n=57) and 112 completed the study. Loteprednol induction therapy 2 weeks before the initiation of long-term tCsA treatment for chronic dry eye disease provided more rapid relief of dry eye signs and symptoms with greater efficacy than tCsA and AT alone [36]. Corticosteroids have proven to be highly effective in the treatment of severe KCS, but patients must be monitored for steroid-related complications such as glaucoma [37].
Voclosporin, a novel CsA analog in ophthalmic solution, is being compared to CsA 0.05% (Restasis®) in subjects with KCS and trial to evaluate the ophthalmic formulation of tacrolimus 0.03% vs. CsA started patient enrollment in March 2019 [38].
A number of lubricant eye drops have been the focus of interest to treat DED. A trial comparing a preservative-free lipid-enhanced flaxseed lubricant (Refresh®) to topical CsA emulsion 0.05% for moderate to severe dry eyes was conducted between 2007-2008. Eye drops with nano size droplets technology (Systane®) used in combination with CsA ophthalmic solution at 0.05% were also studied at the around the same time. These two lubricant formulations are available over the counter [39,40].
A randomized comparative study of autologous serum eye drops at 50% concentration vs. preservative free artificial eye drops plus 0.05 % CsA ophthalmic emulsion was also initiated in 2015. This approach is costly but there are reports that support its efficacy [31]. An exploratory study on the use of finger-prick autologous blood for persistent epithelial defects and severe dry eye disease is also underway. In this study, the patient’s whole blood is applied to the eye from a cleaned finger, preliminary data demonstrated improvement in the symptoms of dry eye with no adverse events reported. This approach presents a simple, cost-effective and possibly more acceptable method for treating dry eye disease.
Other approaches are also being sought to treat DED such as: crosslinked hyaluronic acid and coenzyme Q10 (DEDCO trial) [41].
Essential fatty acids are known to be potent anti-oxidants, and high doses were thought to be beneficial to arrest the progression of KCS. A total of 349 patients who had failed other therapies were randomized to the omega-3 supplements and 186 other subjects were assigned to placebo. Daily doses of omega-3s were 2,000 of eicosapentaenoic acid and 1,000 mg of docosahexaenoic acid. The patients were allowed to use other treatments at their discretion while on the study. Few serious adverse events were seen in both groups, confirming the safety of omega-3 supplements however, no benefit from oral supplementation with fish-derived omega-3 fatty acids was recorded as determined by the patients’ corneal and conjunctival staining scores and tear film break-up time [42,43].
Currently, four clinical trials utilizing Reproxalap, a novel immunemodulating RASP (Reactive Aldehyde Species) inhibitor, are ongoing to treat DED; one study completed phase 2b with the results demonstrating that Reproxalap was effective in multiple dry eye disease symptoms and signs [44].
Forced meibomian gland expression (MGX) in conjunction with intense pulsed light (IPL) is also being evaluated for the treatment of dry eye. Objective measures will include tear cytokine levels, impression cytology, meibography, tear osmolarity and others. Subjective measures will include quality of life screening tools [45].
The results of two clinical trials with Intranasal Tear Neurostimulator are available. A neurostimulator was inserted intranasally which significantly increased acute tear production among the dry eye patients. The first study enrolled 48 patients with Schirmer score of <10mm/5min, and was a randomized, double-masked, crossover, 1-day study in which active intranasal stimulation was compared with two controls: extra-nasal stimulation or sham application. The second single-arm follow-up study had 97 enrollees and lasted for 180 days. The eligibility criteria were similar to those of the first study, except for inclusion of a corneal fluorescein stain score ≥2 in at least one of five corneal regions.
The results of two clinical trials with Intranasal Tear Neurostimulator are available. A neurostimulator was inserted intranasally which significantly increased acute tear production among the dry eye patients. The first study enrolled 48 patients with Schirmer score of <10mm/5min, and was a randomized, double-masked, crossover, 1-day study in which active intranasal stimulation was compared with two controls: extra-nasal stimulation or sham application. The second single-arm follow-up study had 97 enrollees and lasted for 180 days. The eligibility criteria were similar to those of the first study, except for inclusion of a corneal fluorescein stain score ≥2 in at least one of five corneal regions.
Schirmer scores rose with active intranasal application twice daily compared with controls (extra-nasal stimulation or sham application) in the open-label study, and compared with pre-stimulation Schirmer scores in the follow-up study. Patient education was required to apply the neurostimulator in the correct intra-nasal position [46].
Among the DMARDs, hydroxychloroquine provides a significant improvement in patients with dry eyes [47]. While the choice of antiinflammatory drugs remains debatable, it is becoming increasingly clear that most patients with KCS would benefit from some form of anti-inflammatory therapy.
3. Episcleritis
Episcleritis is due to episcleral inflammation leading to acute redness, photophobia and mild discomfort in the eye [2]. See Figure 1.
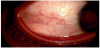
3.1 Epidemiology
The second most common ocular manifestation of RA is episcleritis. The prevalence of episcleritis in patients with RA in the literature ranges from 1-5% [3,48]. Episcleritis may occur at any stage of RA. According to one study, episcleritis occurred at a rate of 5.06% in patients with either stages I (initial inflammation) or II (moderate pain with early evidence of joint damage) RA [2]. There are two major subtypes of episcleritis: Nodular episcleritis is confined to a limited portion of the episclera, while diffuse episcleritis involves a larger contiguous episcleral surface, affecting about 30% and 70% of the patients respectively [2]. About 80% of all cases of episcleritis are limited to one eye, but in patients with systemic diseases such as RA the incidence of bilateral eye involvement is higher [2]. Overall incidence of episcleritis in the general population is less than 1/1000, with a female predominance [2]. The incidence of episcleritis in women between the ages of 45-64 was found to be 74 in 100,000 while the overall incidence in the general population was only 40.7 in 100,000 [2].
3.2 Pathogenesis
Episcleritis (also known as sub-conjunctivitis) is inflammation of the episclera. The sclera comprises the majority of the outer rim of the eye. It is the opaque white part of the eye that starts from the limbus (the region next to the cornea) and extends all the way back to the optic foramen [49]. The episclera is composed of three major parts; the innermost layer is the lamina fusca, which blends with the suprachoroidal and supraciliary lamina of the uveal tract; the middle layer is the lamina proper which is avascular and composed of dense bundles of collagen fibers, and the outer layer is the episclera, which is a thin collagen layer, very vascular containing fibroblast, macrophages and lymphocytes [49]. Surrounding the episclera is the conjunctiva which is clear tissue [49]. The episclera receives vascular supply from the anterior ciliary arteries [49]. These blood vessels are normally hidden, but during inflammation they become congested and prominent and can be easily seen using slit lamp microscopy [23]. Episcleritis results from non-granulomatous inflammation of the superficial vasculature of the episclera, leading to infiltration of white blood cells and vasculature engorgement [50]. The underlying causes of the inflammatory response remain unknown, but it is possible that autoimmune disease and/or infections may serve as triggers. It has been demonstrated that healthy tears contain several anti-inflammatory cytokines such as transforming growth factor- β (TGF- β) and IL-1Ra [51,52]. It has also been demonstrated that systemic inflammation alters the levels of these cytokines [53]. As such, episcleritis is likely compounded by systemic inflammatory signals spilling into the tear fluid and leading to a loss of tear fluid integrity. Ultimately this dysregulation can lead to pathology such as episcleritis. See Figure 2.

3.3 Diagnosis
The clinical presentation of episcleritis may vary, but in general patients present with tearing, mild pain, tearing, irritation, and redness limited to one quadrant of the eye. This is in contrast with scleritis, which usually affects multiple quadrants of the eye, and presents with more severe symptoms such as discharge, photophobia, and decreased visual acuity [54]. Typically, a diagnosis of episcleritis can be made clinically, there are no published guidelines for establishing a diagnosis. However, because scleritis has the potential for sightthreatening sequelae, it is necessary to distinguish between scleritis and episcleritis. Administering phenylephrine to the affected eye and evaluating the vasculature can accomplish this. Because only a few blood vessels pass through the sclera, it is considered “avascular”, while the episclera is considered highly vascular. Therefore, episcleral vessels blanch and scleral vessels do not blanch with phenylephrine. Thus, if inflammation is limited to the episclera then administration of phenylephrine will lead to a relatively normal appearing eye [54]. Patients with an underlying systemic disease such as RA may have a higher incidence of episcleritis than that reported in the literature, because the treatment of moderate-severe episcleritis is systemic anti-inflammatory agents and/or topical steroids and most RA patients are already receiving these therapies. Therefore, the symptoms of episcleritis may be either prevented or masked from clinical presentation. In circumstances where RA patients present with complaints of eye irritation or redness, a fundoscopic exam and referral to an ophthalmologist is in order. If episcleritis is the initial presentation in an otherwise healthy individual, it is recommended that an auto-immune work-up be performed to unveil the underlying systemic inflammatory condition.
3.4 Management
Episcleritis is a benign condition, and usually self-limited without long-term complications. Most cases resolve within a few weeks. Unfortunately, in patients with underlying systemic diseases such as RA, there is a higher rate of recurrence. In such patients, the best management is to treat the underlying disease. Topical antiinflammatory medications such as NSAIDs have been tried in the past, but with limited success [55]. Rather, artificial tears have proven to better control symptomatology. In patients with moderate to severe or refractory symptoms topical corticosteroids are highly effective. It is recommended that patients be placed on low potency steroids first, and if symptoms do not resolve, then move to higher potency steroids such as prednisolone acetate. Patients should be re-evaluated every 2-3 weeks when using steroids, and steroids should be tapered once symptoms resolve. Only in extremely rare situations are oral steroids recommended. The major drawback to the use of steroids is the risk of increasing intraocular pressure (IOP) and cataracts [56]. If the IOP is well monitored, then topical steroids is the preferred route to control inflammation.
No clinical trials have been conducted for episcleritis.
4. Scleritis
Scleritis is, a painful condition that can lead to visual impairment, characterized by the inflammation of the sclera, or the white, outer layer of the globe. It can be further categorized by the anatomical location as either anterior or posterior. Anterior scleritis is defined as the inflammation anterior to the insertion site of the extraocular recti muscles [49]. Posterior scleritis is defined as inflammation of the sclera posterior to the insertion site of the recti muscles. Anterior and posterior scleritis can be distinguished clinically [49]. Anterior scleritis will display signs of anterior segment inflammation such as a visibly injected sclera, keratitis, or cell and flare in the anterior chamber. Posterior scleritis can manifest as retinal findings such as chorioretinal granulomas, serous retinal detachment, optic nerve edema, or a T-sign as seen on B-scan ultrasound [57]. See Figure 3.
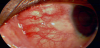
Anterior scleritis can be further divided into subtypes, which include necrotizing scleritis, and non-necrotizing scleritis. Nonnecrotizing scleritis can be either diffuse or nodular [58].
These specific subdivisions have clinical and management implications. Necrotizing features also suggest that there is a component of vessel compromise or vasculitis. As such a more focused rheumatologic assessment should be pursued in anterior necrotizing scleritis as several rheumatological diseases, including RA, can have features of vasculitis [59]. Another distinct entity that can be seen among RA patients is scleromalacia perforans, which is a rare entity seen in roughly 1-3% of patients with scleritis. It is a form of non-inflammatory necrotizing anterior scleritis that is painless, gradual in onset, occurring in patients with long-standing RA usually more than 10 years [2,60]. Ocular complications of scleritis include cataract, glaucoma, and exudative retinal detachment, all potentially capable of impairing vision [56].
4.1 Epidemiology
The prevalence of scleritis is 6 in 100,000 in the general population. Prevalence has been shown to be 1-6% in patients with RA, and up to 7% in patients with granulomatosis with polyangiitis [61]. Scleritis affect women twice as often as men, and typically appears between the ages 30 and 50 [61]. Of the subtypes of anterior scleritis, diffuse and nodular non-necrotizing type are the most common [62]. Necrotizing scleritis makes up to 5% of scleritis cases and accounts for a significant proportion of impaired vision associated with systemic disease. Fifty percent of the necrotizing scleritis cases involve impaired vision, and 80% of necrotizing scleritis cases have an associated systemic disease [57]. Since posterior scleritis presents quite variably, it is difficult to diagnose and is the rarest of scleritis subtypes [63].
It is reported that between 22-42% of patients with scleritis have a systemic autoimmune disease. Of these patients with systemic autoimmune disease, 9.9% are found to have specifically RA [57]. Roughly 23% of those patients with scleritis and an autoimmune disease initially presenting for scleritis were subsequently diagnosed with a systemic autoimmune disease [58]. Scleritis tends to manifest among patients with long standing autoimmune disease including RA [58].
4.2 Pathogenesis
There is no consensus on a linear pathogenesis of scleritis in RA however, the biopsies of a diverse group of scleritis patients revealed lymphocytes predominantly [64]. Studies conducted utilizing biopsies of necrotizing scleritis demonstrated T-cells expressing IL-1 and TNF-α, a culprit cytokine involved in the pathogenesis of rheumatoid arthritis [65,66]. As such it has been postulated that TNF-α, could be an important cytokine in scleritis, given that TNF-α has been demonstrated to increase the activation of MMP in in-vitro scleral fibroblasts. The implications of MMP activation are many, but include allowing a pathway for immune cells to infiltrate through the sclera [67]. A case series identified three subgroups of scleral inflammation [67]. Scleral inflammation from infectious etiology, concomitant with systemic autoimmune disease, and lastly, idiopathic in nature [67]. Granulomatous inflammation with focal tissue necrosis is often found in patients with systemic autoimmune disease [68]. Chronic inflammatory infiltrate consisting of macrophages, B and T cells without necrosis is found in patients with idiopathic scleritis [68]. In contrast, other reports describe that in patients with scleritis and systemic autoimmune disease, the scleral tissue demonstrated inflammatory infiltrate of B cells and macrophages without features of vasculitis [69]. The vasculitis feature is important because vasculitis
is thought to contribute to the focal necrosis via immune-complex deposition. The conflicting information is not entirely surprising given that the features of vasculitis are classically seen in patients who are also anti-neutrophilic cytoplasmic antibodies (ANCA) positive rather than in patients who may have rheumatoid arthritis exclusively [59,68,70]. Moreover, there has also been work done in mice models of inflammatory eye disease (specifically Experimental Autoimmune Uveoretinitis model for uveitis as well as scleritis) which hypothesize that a T helper cell 17 (Th17) response may be culprit in triggering the TNF-α activity in the eye [71]. In patients with severe necrotizing scleritis, features of vasculitis have been found, including immune complex deposition. It has been proposed that the mechanism of scleral wall destruction MMPs. In patients with severe necrotizing scleritis, scleral fibroblasts, local macrophages and T-cells have been shown to express MMP-3, MMP-9 and tissue inhibitors of metalloproteinases (TIMPs), as well TNF-α [62,72]. In scleral fibroblasts, IL-1 and TNF-α were shown to induce the production of MMP-3 and MMP-972. See Figure 2.
4.3 Diagnosis
Patient’s complaints include red eye and severe pain that awakens the patient from sleep. The eye pain can be described as a sensation of fullness to sharp and stabbing in character. On exam, congested vessels that do not blanch with phenylephrine will be seen in conjunctiva, sclera and episclera; globe tenderness, diffuse scleral redness with a bluish discoloration, that points to thinning of the scleral structure, and scleral nodules [73,74]. See Figures 4 and 5. In anterior scleritis, the slit exam can reveal conjunctival injection, ulceration of the conjunctiva and sclera and scleral thinning. B scan ultrasonography demonstrates diffuse thickening of the choroid, sclera and episcleral tissues. The T-sign seen in B-scan ocular sonography is the result of fluid beneath the Tenon’s capsule that creates a squaring off of the interface between the optic nerve and the sclera [42].
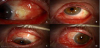
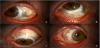
4.4 Management
The management of mild non-necrotizing anterior scleritis starts with the use of oral non-steroidal anti-inflammatory agents such as either selective or non-selective cyclooxygenase (COX) inhibitors without topical steroids [75-76]. Given that biopsies of severe necrotizing scleritis, have shown features of localized vasculitis, patients with severe scleritis are treated with systemic steroids and immunosuppressive agents such as methotrexate, mycophenolate mofetil, cyclosporine, and cyclophosphamide. The decision to consider steroid sparing agents for therapy, appears to be based on the duration of symptoms. With symptoms lasting longer than 1 month, it is advisable to start steroid-sparing agents [77]. It has been shown that biological agents such as TNF inhibitors and anti-B-cell antibodies, such as rituximab, can be utilized to induce disease remission as we will discuss in the next section [78]. Moreover, in non-necrotizing anterior scleritis, subconjunctival steroid injections have been shown to be effective [79].
4.5 Clinical trials impacting management of scleritis
The clinical trials for scleral inflammation utilize the following grade scale after the application of 10% phenylephrine drops, 0 (no scleral inflammation with complete blanching of vessels), 0.5+ (minimal/trace inflammation with localized pink appearance of the sclera around minimally dilated deep episcleral vessels), 1+ (mild inflammation with diffuse pink appearance of the sclera around mildly dilated deep episcleral vessels), 2+ (moderate inflammation with purplish pink appearance of the sclera with tortuous and engorged deep episcleral vessels), 3+ (severe inflammation with diffuse significant redness of sclera, the details of superficial and deep episcleral vessels can't be observed), and 4+ (necrotizing inflammation with diffuse redness of the sclera with scleral thinning and uveal show).
A total of 17 studies are registered for scleritis.
Sirolimus sub-conjunctival injections for autoimmune scleritis, was a phase 1/2, single-center, open-label, non-randomized, prospective pilot study. Sirolimus is a macrocyclic lactone antibiotic with immunosuppressive properties that leads to inactivation of T cells. Sirolimus binds to the same intracellular receptor as tacrolimus and cyclosporine, but does not inhibit calcineurin, but rather blocks the “mammalian target of rapamycin” (mTOR) which interrupts signaling pathways for several cytokines and growth factors including IL-2. Five participants with scleral inflammatory grade of ≥1+ received a baseline injection. The primary outcome was a 2-step reduction or reduction to zero in scleral inflammation by eight weeks. All participants met the primary outcome by week 8 however, 3/5 patients experienced flares (60%) requiring re-injection. No systemic toxicities were observed. Two participants (40%) experienced a localized sterile inflammatory reaction at the site of the injection which resolved without complication [80].
Gevokizumab for active scleritis, was a phase 1/2, open label, non-randomized, prospective, single-arm, pilot trial to evaluate gevokizumab, an IL-1β inhibitor. A decrease of 2 points in scleral inflammation or reduction to zero was the primary outcome. Subcutaneous injections of 60 mg gevokizumab were given at baseline and weeks 4, 8 and 12. Eligible patients could be enrolled in the first extension (up to 36 weeks) and followed-up to 52 weeks. Eight patients with >1+ scleritis were enrolled. Seven eyes from 7 patients met the primary outcome within a median time of 2 weeks following the first gevokizumab injection, no changes in visual acuity or intraocular pressure were noted. There were no serious adverse events related to the study drug. The results of this pilot trial suggested that gevokizumab was well tolerated and it may be beneficial in treating active, noninfectious anterior scleritis [81].
Infliximab has also been used to treat scleral inflammation associated with autoimmune disease. A case series of 10 patients (4 had RA) with refractory scleritis received infliximab infusions with a favorable response seen in 90% of the cases, with 60% of them achieving remission and cessation of concomitant immunosuppression [82].
A multicenter randomized open-label trial Comparing the Efficacy and Safety of Cyclophosphamide vs. Infliximab in subjects with Idiopathic Refractory Scleritis (CIRIS) was initiated in Lyon, France in July 2019. One study arm will receive prednisone, infliximab and low dose methotrexate for 16 weeks and the second arm, prednisone and cyclophosphamide 700mg/m2 every 4 weeks for 16 weeks. Sought outcomes are a decrease in scleral inflammation of two points by week 20, frequency of relapse, time of relapse and the characteristics of the scleritis at worsening [83].
The first randomized, double-masked, dose-finding clinical trial of iontophoretic administration of dexamethasone phosphate for scleritis enrolled 18 patients. The treatment was well tolerated and safe, with less efficacy seen in the lowest (1.2 mA/min at 0.4 mA) dose group, with 5/7 eyes meeting the primary efficacy outcome within 28 days. Iontophoretic delivery of corticosteroids appeared to be a promising treatment for scleritis [84].
Rituximab, a monoclonal antibody against the B-lymphocyte antigen CD20, was also studied in the treatment of refractory scleritis. Twelve patients with noninfectious scleritis refractory to systemic corticosteroid and ≥1 other systemic immunosuppressive were enrolled to receive Rituximab in a prospective, dose-ranging, randomized, double-masked phase I/II clinical trial. Rituximab was given on days 1 and 15 at doses of either 500 mg or 1000 mg. Initial responders with recurrent inflammation were permitted two open-label 1000-mg rituximab infusions after week 24. Nine patients met the scleritis endpoint (2 point-reduction) at 6 months and 4 additionally were able to reduce corticosteroid dose by ≥50%. Seven required re-infusion to maintain inflammatory control. Rituximab was well-tolerated however, the patients who responded to rituximab did not have RA-associated scleritis rather they had granulomatous polyangiitis therefore, the efficacy of rituximab in scleritis due to RA is not fully elucidated [85].
Tofacitinib, a selective inhibitor of the Janus kinase family (JAK) inhibits JAK 1, JAK2 and JAK3. A phase 2 intervention model utilizing 11mg of tofacitinib was initiated in January 2019 to treat inflammatory eye disease including scleritis and/or uveitis [86].
Other clinical trials on scleritis are focusing on gaining further knowledge on the disease prevalence among patients with autoimmune disease and the evaluation of diagnostic modalities such as optical coherence tomography.
5. Peripheral Ulcerative Keratitis (PUK)
5.1 Epidemiology
Peripheral ulcerative keratitis (PUK) can be described as the inflammation of the peripheral cornea, resulting in epithelial defects, potentially leading to perforation of the cornea and blindness. See Figure 6. PUK is often associated with systemic disease such as RA and systemic vasculitis syndromes including ANCA-associated vasculitis, polyarteritis nodosa, relapsing polychondritis, and systemic lupus erythematous [57]. PUK is relatively uncommon with an incidence of 3 cases per million per year [87]. An epidemiologic study of PUK encountered the association with systemic disease in 35.5% of cases, with long standing RA being the most common [88]. Unilateral disease occurs in 83.6% of cases. PUK has a male predominance (60%) with a third of patients reporting previous ocular surgery in the affected eye [89]. For patients with known RA, the mean disease duration was 20 years [87]. It is clinically relevant to know that once inflammation begins within the corneal stroma, loss of visual acuity can occur within a few days [88].
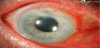
5.2 Pathogenesis
The cornea is comprised of five layers. The outer layer is the epithelium, followed by the Bowman’s membrane, the stroma, the Descemet’s membrane and the inner epithelium. The stroma is thickest at the periphery and is primarily made of collagen. The peripheral cornea receives some of its nutrients from nearby capillary beds, which may explain its susceptibility to inflammatory infiltrate [88,90]. In PUK, inflammatory cells accumulate in the periphery of the corneal stroma, adjacent to the corneal limbus. The inflammation forms a crescent shape, resulting in a peripheral epithelial erosion and stromal thinning. The predominantly peripheral involvement suggests that at least in part the pathogenesis is due to immune complex deposition rather than solely based on cellular-immunity. This postulate is based on the fact that the central cornea has no blood supply and remains unaffected by PUK [91]. PUK may lead to perforation of the cornea or corneal melt syndrome, which can result in blindness [92]. In RA-associated PUK, one emerging hypothesis suggests that CD4+ T-cell activation leads to antibody production. The antibodies then form immune complexes that deposit in the corneal margin [93]. Activation of the complement system then leads to the migration or chemotaxis of activated neutrophils and macrophages, which then release destructive collagenase and proteases such as MMP-1, MMP-2 and MMP-9 [91,94,95]. Similarly to scleritis, high TNF-α levels have been described in PUK which is consistent with RA pathogenesis [95].
5.3 Diagnosis
PUK presents as a painful, red eye and may be associated with tearing and blurry vision. On slit lamp exam, the epithelium is visibly absent from the crescent-shaped corneal ulcer. Cellular infiltrate may be seen in the stroma in addition to, stromal thinning and secondary descemetocele. Inflammation is typically seen in the adjacent conjunctiva and sclera. Although PUK is often isolated, PUK has been observed simultaneously with scleritis in 36% of cases [96].
Diagnosis is based on history and physical exam. Evaluation for systemic underlying disease should include antinuclear antibodies, ANCA, rheumatoid factor, anti-citrullinated peptide antibodies (ACPA), rapid plasma reagin (RPR), fluorescent treponemal antibodies, interferon-γ release assay and purified protein derivative (PPD) for tuberculosis, hepatitis B and C virus antibodies. B-scan ultrasound can evaluate for concurrent posterior scleritis. Further workup to assess for localized infectious etiology can include corneal scraping for culture [91]. If necessary, tissue biopsy of the lesion can assist in diagnosis, and to evaluate for additional systemic disease [91].
5.4 Management
Management strategies for PUK are based on disease etiology, severity, and the presence of underlying systemic disease. Ocular treatment with preservative-free artificial tears and bandage contact lenses helps promote epithelialization of the ulcer, and topical antibiotics use helps prevent bacterial superinfection. In noninfectious PUK, systemic glucocorticoids can be administered and tapered based on the clinical response, especially in severe disease to minimize the risk of corneal perforation [88]. Additionally, systemic immunosuppressive agents can significantly reduce the progression of ocular disease, improve or stabilize the visual acuity, and prevent the development of the extraocular disease [93]. Biologics, such as rituximab, infliximab, and adalimumab, can be effective in cases refractory to initial corticosteroid and immunosuppressive management [92,97].
Surgical management of PUK in severe cases includes tissue adhesive bandage contact, lamellar graft, and tectonic corneal grafting. Further, while resection of adjacent conjunctival tissue has been proposed as a potential method to reduce vascular access to the affected area by preventing inflammatory cells from migrating to the cornea, [98] but this approach still requires further evaluation and validation. No clinical trials are available for PUK.
6. Anterior Uveitis
6.1 Introduction
The uvea is anatomically defined as the middle layer of the eye, identifiable between the sclera and the retina. Anterior uveitis encompasses inflammation of any or both of the components of the anterior uvea. Inflammation of the iris alone is termed iritis, and inflammation of both the iris and the ciliary body is iridocyclitis. Posterior uveal inflammation defines choroiditis, inflammation of the vascular tissue adjacent to the retina (choroiditis). For this section of the review, we will focus on anterior uveitis. See Figure 7.

6.2 Epidemiology
Inflammatory ocular disease (IOD) among RA patients presents with anterior uveitis in 14-42% of the cases. Given the retrospective nature of the studies, uveitis definition (anterior vs. posterior) [99] and small sample size led to lower percentages of anterior uveitis prevalence among RA patients [100]. However, a nine year retrospective study identified collagen vascular disease in 18% of the patients with uveitis and within this group RA diagnosis was ascertained in 42% of the cases with anterior uveitis [101].
6.3 Pathogenesis
The anterior eye is an immune privileged environment. The immunosuppressive microenvironment drives apoptosis of immune cells through constitutive expression of Fas ligand or CD95L (FasL) within the eye. During the early stages of uveitis, immunopotentiating cytokines may be released into the anterior chamber, leading to elimination of immunosuppression and the development of inflammatory/autoimmune ocular disease [102]. While a specific disease mechanism linking RA and anterior uveitis has not been determined [103], studies have defined that signal transducer and activator of transcription 3 (STAT3), a transcription factor, can favor to the expansion of Th17 T cell functions and plays a role in development of experimental autoimmune uveitis (EAU). A wildtype, micro RNA 155 (miR-155-/-), and CD4-STAT3KO mice demonstrated that STAT3 is required for miR-155 expression, and that STAT3-dependent increase in miR-155 expression in vivo correlated with onset of EAU. Furthermore, miR-155 -/- and CD4- STAT3KO mice did not suffer from EAU [103]. Since Th17 expansion and STAT3 have been implicated in RA as well [104], it stands that this pathway may play an underlying role in patients with concurrent anterior uveitis and RA.
Beyond the specific pathological actions of T-cells in the usually immune privileged anterior chamber space, MMPs have also been implicated in both RA and anterior uveitis disease. The aqueous humor is the most accessible form of the ocular microenvironment, and naturally holds soluble factors that inhibit activation/proliferation of lymphocytes [102]. Hence, it has been postulated that in anterior uveitis disease, changes in the consistency of the aqueous humor occur first, followed by lymphocyte infiltration which drives further disease. To confirm this hypothesis, measurements of the levels of MMPs in the aqueous humor of patients with and without uveitis have been compared; at the same time the MMP levels in the aqueous humor in animal models of Lipopolysaccharide (LPS)-induced uveitis [72] were found to increase after LPS injection, demonstrating a correlation between MMPs in the aqueous humor and anterior uveitis [72]. Furthermore, MMPs have been implicated in the development of arthritic disease, enforcing the notion that, this cytokine may be the underlying driver for anterior uveitis in RA patients.
6.4 Clinical diagnosis
Anterior uveitis is a clinical diagnosis often defined by pain, decreased vision, synechiae, tearing, photophobia, and eye redness without discharge. Anterior uveitis occurs in multiple clinical syndromes, including infectious uveitis and non-infectious uveitis. Non-infectious uveitis can include autoimmune disease associated uveitis however, autoimmune origin of inflammatory ocular disease (IOD) only encompasses 5% of cases of uveitis and scleritis [105]. For the purposes of clinical diagnosis, only a small subset of the cases of uveitis will be due to autoimmune disease, and an even smaller subset will be due to RA. A retrospective study conducted in an eye clinic, found that 43.4% of the patients had ocular inflammation and an autoimmune disease but that only 2% of the patients had the ophthalmic manifestation as the only expression of the autoimmune disease. In rare cases, the ocular disease preceded the RA diagnosis [100] highlighting the need to consider the clinical diagnosis of RAassociated anterior uveitis. Most cases of anterior uveitis are seen among known RA patients. However, both the rheumatologist and the ophthalmologist should be weary that ocular symptoms can be the first manifestation of an autoimmune disease.
When considering ophthalmic disease associated with RA, ACPA has been demonstrated to be a specific serologic marker. While rheumatoid factor (RF) can be found in many autoimmune diseases, ACPA, when present, can be greater than 95% specific for RA [105]. For ophthalmic disease in particular, uveitis and dacryoadenitis should trigger a thorough history and physical exam with special attention to the review of systems, as well as laboratory and hand imaging testing looking for periarticular osteopenia, joint space narrowing [106]. In cases of new uveitis diagnosis, investigations should include syphilis testing; erythrocyte sedimentation rate and C reactive protein should also be obtained as could be the clues to uveitis associated with rheumatic diseases [101]. If joint symptoms are concurrently observed, then Lyme disease, ANCA, RF, and ACPA serologies should be obtained.
One additional consideration should be the evaluation of the posterior uveal space, by assessing the thickness of the choroid. RA patients with decreased choroidal thickness who received hydroxychloroquine were found to have improvement with increase choroidal thickness on follow-up [107].
6.5 Management
The majority of cases of anterior uveitis, including those associated with RA, can be successfully treated with topical glucocorticoids. Topical cycloplegic/mydriatic medications to dilate the pupil can also be used [99]. Topical non-steroidal anti-inflammatory drugs have also been reported with few patients requiring oral glucocorticoids for treatment [99].
However, the supplemental utilization of immunosuppressive therapy in patients with uveitis and autoimmune disease, such as RA, should be considered early in the treatment course because cases of uveitis associated with rheumatic disease have a significantly increased response to immunotherapy101. Cyclosporine [101], anti-TNF-α agents [108], and tocilizumab [108] have been utilized. Tocilizumab and other IL-6 modulators deserve special consideration in cases of autoimmune associated uveitis [109], as opposed to reserving its use for refractory cases [108].
7. Clinical Trials Impacting the Management of Uveitis
A total of 45 studies were initiated to study anterior uveitis however, none of them focused on RA-associated anterior uveitis. The grade of cell count, flare, limbal injection, presence of hypopion, precipitates and anterior synechia by slit lamp exam of the anterior chamber are utilized to score the severity of the anterior uveitis.
We will review some of the most relevant studies on anterior uveitis.
The FDA approval of adalimumab a TNF-α blocker, for noninfectious intermediate, posterior and panuveitis was based on two randomized, double-masked, placebo-controlled studies (UV I and II) utilizing adalimumab (Humira®) at an initial dose of 80 mg followed by 40 mg every other week. The primary endpoint in the studies was time to treatment failure. Study UV I enrolled 217 patients with active uveitis while being treated with oral prednisone. Prednisone 60 mg/day was given at study entry followed by a mandatory taper and discontinuation by week 15. Study UV II evaluated 226 patients with inactive uveitis while being treated with corticosteroids to control their disease. Prednisone 60 mg/day was given at study entry, followed by tapering, with complete corticosteroid discontinuation by week 19. Results from both studies demonstrated statistically significant reduction of the risk of treatment failure in patients treated with adalimumab versus patients receiving placebo. Study UV I: adalimumab (54.5%) versus placebo (78.5%). Study UV II: adalimumab (39.1%) versus placebo (55%) [110,111].
Golimumab approved in 2009 for ankylosing spondylitis was noted to be associated with significantly less episodes of anterior uveitis. The occurrence rate of uveitis before and after initiation of golimumab was studied on 93 patients in the Netherlands. The rate of uveitis, during the year after golimumab was initiated, was compared to the uveitis rate in the 12 month before the treatment and the rate of anterior uveitis decreased from 11.1 per 100 patient-years to 2.2 per 100 patient-years (RR = 0.2; 95% CI, 0.040.91) during golimumab treatment [112].
The SOLACE trial to evaluate the efficacy of reproxalap in noninfectious anterior uveitis was discontinued after no significant improvements were found in the treatment arm compared to vehicle [113].
A clinical trial of Tocilizumab, an IL-6 receptor antibody, for refractory uveitis in juvenile idiopathic arthritis (JIA) was initiated in 2018. The multicenter investigator-initiated, single arm phase 2 trial was conducted in the United Kingdom; 22 children with active JIA-associated uveitis, who had failed to respond to MTX and anti- TNF agent were enrolled. All participants were treated for 6 months, with follow-up of 9 months from registration. MTX at a stable dose and 162 mg of Tocilizumab every 2 weeks (weight >30kg) were given. At 12 weeks of treatment (primary outcome), 11 children had some improvement in uveitis and 7 of them had achieved a two-step decrease in the level of inflammation as measured by the standard criteria [114,115].
8. Conclusion
General practitioners and rheumatologists should be aware of the association of RA and anterior chamber ocular disease, particularly considering KCS, episcleritis, scleritis, PUK, and anterior uveitis when RA patients present with ocular symptoms ranging from grittiness and mild discomfort to new redness, pain, vision changes and inflammatory ocular disease.
Putative pathophysiologic mechanisms for ocular disease in RA, include oxidative stress, cytokine imbalance, chronic inflammation, vascular permeability, immune complex deposition and the role of T-cells as well as the contribution of tear hyperosmolarity among other factors.
Ocular symptoms can be the initial presentation of RA or other autoimmune disease so a complete evaluation is appropriate for cases without established RA diagnosis. Once the autoimmune nature of the ocular disease is suspected, a multidisciplinary approach will prove to be the best option to manage RA-associated ocular pathology.
KCS, episcleritis, scleritis, and PUK, necrosis, iritis or iridocyclitis usually necessitate the use for intensive systemic immunosuppressive therapy. The clinical trials have been pivotal in changing the management of inflammatory eye disease in autoimmune diseases. Options include biologics, immunomodulators, autologous serum tears, aldehyde trap inhibitors, as well as novel mechanisms of drug delivery including nanomicellar formulation and iontophoresis. Screening and timely recognition and intervention improves the prognosis of the ocular manifestations of RA.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
We are grateful to Dr. Thomas A. Oetting, Eye.Rounds.org University of Iowa for figures 4, 5 and 6 and to Mrs. Andrea Rose- Holder for her assistance in the preparation of this manuscript.