


1. Introduction
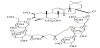
Steamed and dried tubers of Bolbostemma paniculatum (MAXIM.) FRANQUET (Cucurbitaceae), named as "Tu Bei Mu", has been used as an anti-inflammatory agent for mastitis and an antidote for snake skin in China, and tubeimoside-1 (T-1), a cyclic bisdesmoside, was isolated as one of active ingredients from the methanolic extract of the tubers (Figure 1) [1,2]. T-1 suppresses cell growth and induces cell apoptosis of various cancer cells in vitro [3,4]. T-1, an amphiphilic compound having a relatively lipophilic exterior surface and nonpolar interior cavity, increased the solubility of poorly water soluble compounds such as Saponin A, Yellow OB and α-tocopherol [1,5]. In the present study, using T-1, the solubilizing activity to itraconazole (ITZ), effect on oral absorption of ITZ in rats, stability in aqueous buffer and intestinal homogenate, cytotoxicity to Caco-2 cells, and membrane permeability across rat small intestine and Caco-2 cell monolayers were examined to understand the biopharmaceutical properties of unique amphipathic inclusion compound and to search for the usefulness of T-1. Itraconazole (ITZ) and hydroxypropyl-β- cyclodextrine (HPβCD) were used as a poor water-soluble compound and a potent inclusion compound, respectively [6]. As clinically available formulation of ITZ, ITZ (10 mg/mL) is solubilized by 40% HPβCD to form a molecular inclusion complex (SPORANOX® (itraconazole) Oral Solution, Janssen Pharmaceutica N.V. Beerse, Belgium).
2. Material and Methods
2.1 Materials
ITZ and HPβCD were purchased from Sigma Chemical Co. Ltd (St Louis, USA). Cell culture medium and reagents were from Gibco Laboratories (Life Technologies Inc., Grand Island, NY). All other chemicals used were of the highest purity available.
2.2 Extraction of T-1
T-1 was extracted from traditional Chinese herbal medicine Bolbostemma paniculatum (Maxim.) Franquet (Cucurbitaceae) using a methanol extract of the tubers in the same manner as reported previously [1,2].
2.3 Effects of T-1 and HPβCD on ITZ solubility
Excess amount of ITZ (approximately 12 mg) was dissolved in a mixture of propylene glycol (17.5 μL) and 10 mole/L HCl (4.375 μL) pre-warmed at 40-50°C, according to the method reported previously [7]. T-1 or HPβCD saline solution (674.45 μL) containing 10 mole/L NaOH solution (3.675 μL) were added at room temperature. The final concentration of propylene glycol and pH were 2.5% and approximately neutral, respectively. The concentrations of T-1 and HPβCD were adjusted at 0, 2, 10 or 40 %. For comparison, non-ionic polyethoxylated detergent, Cremophor EL (Sigma-Aldrich Japan. Tokyo), was also used at a concentration of 10%. Prior to the analysis, the final mixture containing undissolved ITZ was passed through a 0.45-mm syringe filter.
2.4 Effects of T-1 and HPβCD on oral absorption of ITZ in rats
Male Sprague–Dawley rats aged 7–9 weeks old were used. The protocol of the experiments was reviewed and approved in advance and experiments with animals were performed in accordance with the Guide for Animal Experimentation from the Committee of Research Facilities for Laboratory Animal Sciences, Hiroshima International University, which is in accordance with the Guidelines for Proper Conduct of Animal Experiments from the Science Council of Japan. The license number of this animal study was AE15-003. As a control, ITZ was suspended in saline (10 mg/mL) containing 2.5% propylene glycol. T-1 or HPβCD was added to the saline suspension of ITZ at a concentration of 10%, in the same manner as described above. One night fasted rats received ITZ suspension by stomach intubation at a dose of ITZ 20 mg/2 mL/kg. Blood (0.25 mL each) was collected from jugular vein periodically under light anesthesia with diethylether and centrifuged to obtain plasma.
2.5 Stability study of T-1
The stability of T-1 in pH 7.4 phosphate buffered saline (PBS: 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4 and 1.5 mM KH2PO4) supplemented with 1 mM CaCl2, 0.5 mM MgCl2 and 5 mM glucose (PBS-G), pH 0.7 HCl solution and S9 fraction of rat intestinal mucosa homogenates was determined at 37°C. The S9 fraction was prepared by centrifuging 10% mucosa homogenate in pH 7.4, 10 mM HEPES-NaOH buffer containing 150 mM KCl and 1 mM phenylmethanesulfonyl fluoride (PMSF) at 9,000 x g for 20 min at 4°C. Concentrations of T-1 and protein in S9 fraction were adjusted at 1 mg/mL and 100 μg/mL, respectively, and the solution was incubated at 37°C. The reaction was stopped by adding excess amount of acetonitrile and cooling to 4°C.
2.6 Cytotoxicity of T-1 to Caco-2 cells
The viability of Caco-2 cells after exposure to T-1 was assessed using WST-1 (2-(4-Iodophenyl)-3- (4-nitrophenyl)-5-(2,4-disulfophenyl)- 2H-tetrazolium, monosodium salt, a cell proliferation reagent) according to the manufacture’s protocol (Dojindo, Kumamoto, Japan). Briefly, Caco-2 cells cultured onto 96-well plates for 21 days were washed with pH 7.4 PBS (100 μL) and treated with 100 μL of T-1 solution (0.1–1,000 μM) dissolved in serum free Dulbecco's Modified Eagle Medium (DMEM). After 1 or 24 h-incubation at 37°C, T-1 solution was discarded and WST-1 reaction mixture (100 μL) containing WST-1 and 1-methoxy phenazinium methylsulfate (1-methoxy PMS) was added. After 60 min-incubation, the UV absorbance at 450 nm was measured with a microplate reader (SpectraMax Plus 384, Molecular Devices Japan). The 50% inhibitory concentration of cell growth (IC50) was calculated by curve fitting.
2.7 Membrane permeability of T-1 across intestinal loop in situ and Caco-2 cell monolayers in vitro
Bile duct of anaesthetized rats was ligated and the intestinal lumen was washed with a sufficient amount of saline prewarmed at 37°C. Three intestinal loops of 10 cm-long each were made by ligating both ends of the intestinal loop at proximal (a segment from 5 cm below the bile duct opening), middle, and distal small intestine (a segment from 5 cm above the ileocecum). T-1 (0.5 mg/mL) was dissolved in pH 7.4 PBS-G. T-1 solution (1 mL) was administered into the loop, and the luminal fluid was recovered 1 h after administration by washing the intestinal lumen with sufficient amounts of PBS-G. The collected luminal fluid was deproteinized with acetonitrile.
Caco-2 cell monolayers cultured in 12-well Transwell® chamber (Costar, Cambridge, MA) were used after monitoring the transepithelial electric resistance (TEER) with a Millicell electrical resistance system testing device (Millipore, Bedford, MA, USA). Monolayers with TEER of more than 500 Ω·cm2 were used. T-1 (10 or 30·μM) was dissolved in pH 6.0 PBS-G. The solution (0.5 mL) was placed on the apical side, and the other side was filled with 1.5 mL of pH 7.4 PBS-G. The Transwell® chamber was incubated at 37°C, and the transport medium (100 μL) in the basolateral side was sampled periodically to determine T-1 concentrations. Fresh transport medium was refilled each time.
2.8 Analysis
Concentrations of ITZ were determined by HPLC. Plasma samples were deproteinized with an equal volume of acetonitril. The column used was YMC-Triart C18 (YMC Co. Ltd., Kyoto, Japan) and the mobile phase was a mixture of acetonitrile and 1% acetic acid (55:45 v/v). The flow rate was 1 mL/min, and detection of ITZ was made at UV 260 nm. Concentration of protein was measured by Bradford method using bovine serum albumin as the standard [8]. Concentrations of T-1 in various samples were measured by using liquid chromatography/mass spectrometry (LC-MS, Shimadzu LCMS8040, Japan) after deproteinization with acetonitrile. CAPCELL PAK C18 MG III S3 (50 mm, 2.0 mm I.D., Shiseido, Japan) was used as a column and eluted with a mixture of 0.25 mM sodium acetate (solution A) and acetonitrile (solution B) using a following linear gradient: 20% to 70% of B for 4 min, 70% of B for 4-4.5 min, and 20% of B for 4.5-8 min. The temperature of the sample cooler was set to 4°C and the flow rate was 0.3 mL/min. Selected ion monitoring (SIM) was performed with the m/z 1341.9. The calibration curve showed a good linear correlation over the concentration range from 0.025 to 10 μM (r = > 0.99).
2.9 Statistical analysis
Data were presented as the mean ± SE. Statistical analysis was performed by Student’s t-test, or by one-way analysis of variance followed by the Turkey-Kramer’s Test for multiple comparisons. A difference of P<0.05 was considered statistically significant.
3. Results
3.1 Effects of T-1 and HPβCD on ITZ solubility
Effects of T-1 and HPβCD on ITZ solubility were evaluated, in which excess amount of ITZ was dissolved in a small volume of a mixture of propylene glycol and HCl, the solution was mixed with 0, 2, 10 and/or 40% T-1 or HPβCD saline solution containing a small amount of NaOH (Figure 2). The final concentration of propylene glycol was 2.5% and pH was neutral. The solubility of ITZ itself was 38.0 μg/mL. In the presence of 10% T-1, the solubility of ITZ increased to 6.47±0.16 mg/mL, and it was about 9-fold higher than that of 10% HPβCD (0.75±0.20 mg/mL). At 40% HPβCD, the solubility of ITZ reached to approximately 10.13±0.14 mg/mL. Cremophor EL, a nonionic polyethoxylated detergent, at a concentration of 10% increased ITZ solubility to 56.5 μg/mL, twice of ITZ alone.

3.2 Effects of T-1 and HPβCD on oral absorption of ITZ in rats
Three different formulations, that is., ITZ saline suspension containing dissolved ITZ (38.0 μg/mL) and undissolved ITZ (9.962 mg/mL), ITZ saline suspension contaning 10% T-1 (ITZ/10%T-1 suspension containing dissolved ITZ (38.0 μg/mL), undissolved ITZ (3.53 mg/mL), and 6.432 mg/mL ITZ/10%T-1 inclusion complex), and ITZ saline suspension containing 10% HPβCD (ITZ/10%HPβCD suspension containing dissolved ITZ (38.0 μg/mL), undissolved ITZ (9.25 mg), and 0.712 mg/mL ITZ/10%T-1 inclusion complex), were administered orally at a dose of 20 mg/2 mL/kg of ITZ (Figure 3). The intestinal absorption of ITZ given as ITZ/10%T-1 suspension was higher than that of ITZ saline suspension as follows: the peak plasma concentration (Cmax) and area under the concentration-time curve from 0 to 4 h (AUC0-4) of ITZ were 0.14±0.03 μg/mL (mean±SE, n= 3-4) and 14.6±3.72 μg•min/mL after saline suspension, and 0.31±0.04 and 46.8±12.0 after ITZ/10%T-1 suspension, respectively. However, Cmax and AUC0-4 of ITZ given as ITZ/10%HPβCD suspension were further increased as follows: 0.78±0.28 μg/mL and 114±30.2 μg•min/ mL, respectively. The AUC0-4 of ITZ/10%HPβCD suspension was significantly higher than that of the saline suspension (P<0.05).

3.3 Recovery of T-1 from intestinal loop in situ
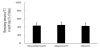
T-1 solution (500 μg/mL) was administered into 10cm long intestinal loops, and the remaining amount of T-1 in the loop 1 h after administration was determined (Figure 4). The recovered amounts of T-1 were comparable among three different regions. In these experiments, T-1 was not detected in blood circulation, suggesting the disappearance of T-1 from the loop was not due to the intestinal absorption of T-1.
4. Discussion
Biopharmaceutical properties of T-1 were evaluated to search for the usefulness of tubeimoside-1, as an unique amphipathic cyclic bisdesmoside. The in vitro solubilizing activity of T-1 has been reported already using several poor water soluble compounds such as Saponin A, Yellow OB, and dl-α-tocopherol [2,5]. However, the efficacy of solubilizing effect of T-1 in oral absorption of poor water soluble compounds in vivo was not yet examined. To compare the solubilizing activity of T-1, HPβCD was used as a reference inclusion compound. HPβCD has high water solubility (more than 60%), possesses high solubilizing activity against variety of poorly water soluble compounds, and is used as a pharmaceutical solubilizer of drugs including ITZ [9,10]. The effect of Cremophor EL, a non-ionic polyethoxylated detergent, on ITZ solubility was also examined for comparison, since T-1 is reported to have a surface tension-reducing activity. The critical micelle concentration (cmc) of T-1 is reported to be 0.001% (7 μM), at which the surface tension of T-1 solution was 54 dyn/cm [5]. ITZ, a triazole antifungal agent, is a weak basic compound with a pKa value of 3.70. The solubility of ITZ, categorized as a biopharmaceutical classification system (BCS) class II compound with low solubility and high permeability [11], is reported to be 1-4 ng/ mL in water and 4–6 μg/mL in pH 1.2 (0.1 N HCl). The dose number (D0 = (maximum dose strength/250 mL) / minimum solubility) of ITZ was estimated to be 800,000, by assuming that the minimum water solubility of ITZ is 1 ng/mL and maximum dose strength is 200 mg each in human. The oral absorption of ITZ is known to be scattered among patients and doses possibly due to the low solubility, and intake of food increases oral bioavailability of ITZ due to the solubilization by bile acids [12,13]. The average value of plasma Cmax of ITZ given orally (200 mg) after fasting was about 59% that after the standard meal in healthy volunteers [13,14]. In contrast, ITZ oral solution containing 40% HPβCD exhibited greater oral bioavailability than ITZ capsule in the fasted state than in the nonfasted state in healthy volunteers [12,15]. Gastric pH alterations due to the coadministration of proton pump inhibitors (PPIs) or acidic beverages such as Coca- Cola are also reported to significantly decrease or increase the bioavailability of ITZ, by modulating the solubility of ITZ in the stomach [16,17]. It is reported that ITZ absorption is promoted by low stomach pH, long gastric retention time and a high fat content of the coadministered meal [18]. The dose-dependent oral bioavailability of ITZ due to the high intestinal first–pass effect in rats is also reported as follows: bioavailability of ITZ was 34.9% at a dose of 10 mg/kg; 63.0% at 30 mg/kg, and 85.7% at 50 mg/kg, in which the hepatic firstpass effect was relatively low [19,20].
In the present study, ITZ was dissolved in a small volume of a mixture of propylene glycol and HCl, and mixed with T-1 or HPβCD solution. The final concentration of propylene glycol was 2.5% and pH of the final mixture was adjusted to approximately neutral. The solubility of ITZ alone was 38 μg/mL, and 10% T-1 increased the ITZ solubility by 170-fold of ITZ alone. The high solubility of ITZ in the present study (38.0 μg/mL) compared to reported value (1-4 ng/ mL) would be due to the presence of propylene glycol (2.5%). The solubilizing activity of T-1 at a concentration of 10% was nine-fold greater than that of HPβCD (Fig. 2). The molar ratio of ITZ (solubility 9.22 mmole/L) against 10% T-1 (75.8 mmole/L) was approximately 0.122, implying that approximately eight T-1 molecules interact with one ITZ molecule. The solubilizing activity of T-1 was not considered to be derived from the surface tension reducing activity, but due to the formation of inclusion complex between T-1 and ITZ, since Cromophor EL (10%), a surfactant, increased ITZ solubility only twice. The absorption enhancing effect of T-1 was examined in rats using ITZ. Though ITZ/10%T-1 suspension increased the AUC0-4 of ITZ three-fold of ITZ saline suspension, ITZ/10% HPβCD suspension further increased the ITZ absorption, more than twice of ITZ/10%T-1 suspension (Figure 3). The reason of the low absorption enhancing effect of T-1 compared to HPβCD, irrespective of the greater solubilizing activity, would be accounted for by the instability of T-1 resulting in the degradation of the inclusion complex between T-1 and ITZ. In loop study, approximately 60% of T-1 was disappeared from the loop within 1 h after dosing, but it was due to the degradation of T-1 in tissues. Also, no membrane permeation of T-1 itself was observed (Figure 4 and Figure 5). In contrast, if the ITZ/10%T-1 inclusion complex was stable in gastrointestinal tract and the solubility of ITZ increased to the level of intact ITZ/10%T-1 inclusion complex, the dose number of ITZ may correspond to 0.12 by adopting 6.47 mg/mL as the minimum solubility of ITZ as well as ITZ/10%T-1 inclusion complex. The low dose number may imply that the intact ITZ/10%T-1 inclusion complex is a high solubility compound with low membrane permeability like BCS Class III compounds. In general, however, the intestinal absorption of compounds having large molecular weights is not expected much, because of the low diffusion coefficients in passing the unstirred water layer (USWL) in the intestine. Most drug/CD inclusion complexes would release the lipophilic drug in the gastrointestinal tract and increase the intestinal absorption of the lipophilic drug. On the other hand, CD molecules that released drug molecules are retained in the intestinal tract. CDs are bulky and hydrophilic compounds and therefore basically poorly absorbed. It is summarized that the oral bioavailabilities of αCD, βCD, γCD, and HPβCD in rats were 2-3%, 0%, less than 0.1%, and less than 3%, respectively [21]. It is also reported that the oral bioavailability of HPβCD in the majority of children patients was less than 1% [22]. It will be important to study further about the releasing mechanism of drug from drug/CDs inclusion complex in the intestinal tract, the releasing timing of drug (before or after diffusion of USWL), the fate of CD molecules after releasing drug molecules, and so on.

T-1 is known to have anti-inflammatory, anti-tumor, and antitumorigenic activities [4,23-26], indicating that T-1 is a cytotoxic compound. A potent cytotoxicity of T-1 was also observed in Caco- 2 cells (Figure 6). Also, the membrane permeability of T-1 across Caco-2 cell monolayers was not detected at a concentration of 10 μM, but T-1 gradually permeated Caco-2 cell monolayers with time at a concentration of 30 μM, possibly due to the cytotoxicity of T-1. Collectively, T-1 was not found to be suitable as a safe solubilizer of low water soluble compounds in enhancing the oral bioavailability in vivo. Instead, T-1 having an anticancer activity may be useful as a drug carrier of other lipophilic anticancer drugs, though some appropriate delivery system such as liposomes for T-1/anticancer agent inclusion complex is necessary.

5. Conclusion
Greater solubilizing activity and biodegradability of T-1, in addition to cytotoxicity, were observed. Further study is necessary to search for the usefulness of T-1 as a strong solubilizer by forming inclusion complex with lipophilic compounds.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
Oda K. designed experimental protocol and performed most of in
vitro and animal experiments.
Umakoshi T. helped Dr. Oda with animal experiments and in vitro
stability study.
Mori N. cultured and performed experiments using cells.
Kasai R. extracted tubeimoside-1 from Tu Bei Mu, and discussed
regarding chemical properties.
Murakami T interpreted data and summarized it as a manuscript.