


1. Introduction
Globe artichoke (Cynaracardunculus var. scolymus L. Fiori) is a perennial plant belonging to the Asteraceae family native to the Mediterranean Basin [1,2]. The edible part is the immature inflorescence that is consumed raw or after some type of thermal treatment. From a nutritional point of view, it is considered as very healthy and nutritious [3,4]. It is a rich source of minerals [5], vitamin C [6], inulin [7] as well as polyphenols [8]. The effect of processing steps on the phytochemical content has also been studied to some extent; an increase of protein and total phenolic content as well as a decrease of dietary fiber content, as a result of boiling, has been reported [9-11]. Fermentation, on the other hand, seems to be an ideal processing step as it increases the level of safety [12,13], improves organoleptic properties [14], enhances nutritional value [15,16] and extends shelf life [17].
Spontaneous fermentation of artichoke immature inflorescence is not that common and therefore no reports in the literature currently exist, at least to our knowledge. Given the nutritional value of artichoke itself and the benefits that fermentation might confer, this type of product is worth studying. Therefore, the aim of the present study was to monitor the physicochemical changes as well as the microbial succession during spontaneous fermentation of artichoke immature inflorescence, by both culture dependent and independent techniques and to taxonomically characterize the dominant microbiota.
2. Material and Methods
2.1 Pickle preparation and sampling
Artichoke fermentation was performed according to a traditional recipe that is used in southern Greece. Immature inflorescences were peeled, cut and submerged into brine solution (8% w/v NaCl) containing lemon juice (one lemon, i.e. 25 mL juice per liter of brine). Blanching at boiling water for 1.5 min took place in the same brine. Then, artichoke pieces were drained and submerged into a new brine solution (8% w/v NaCl) without lemon juice. Olive oil was added on top and left for fermentation for about 26 days at room temperature (20°C). The microbiological quality of the bracts as well as the immature inflorescence before and after treatment with lemon juice and blanching was assessed. During fermentation, brine samples were taken at 0, 1st, 3rd, 5th, 7th, 12th, 19th and 26th day of fermentation. Fermentation was performed twice and the average values are presented.
2.2 Physico-chemical and microbiological analyses
Fermentation was monitored by measuring pH value and total titratable acidity (TTA) in brine samples. Samples (10 g) were aseptically derived from each fermentation jar, and after the pH value was recorded (WTW, Weilheim, Germany) they were homogenized with 90 mL of distilled water using a Stomacher apparatus (Seward, London, UK). The acidity was titrated using 0.1 N NaOH to final pH 8.5. The TTA was expressed in mL NaOH to final pH 8.5. All analyses were performed in triplicate and the average values are presented.
Microbiological analyses were performed in brine samples. Total aerobic mesophilic count, yeasts-molds, lactic acid bacteria, Staphylococcusaureus, enterococci, Bacillus cereus, sulphur-reducing clostridia, Enterobacteriaceae, coliforms and E. coli as well as qualitative and quantitative determination of Listeria monocytogenes and Salmonella were performed according to Paramithiotis et al [18]. More accurately, the pour plating technique was applied for the enumeration of Enterobacteriaceae, coliforms and E. coli. The first was carried out in Violet Red Bile Glucose Agar (VRBGA) (LAB M, Lancashire, UK) and incubation at 37°C for 24 h. Enumeration of coliforms and Escherichia coli was performed in Chromocult coliform agar (Merck, Darmstadt, Germany) and incubation at 37°C for 24 h. The surface spreading technique was performed for the enumeration of total aerobic mesophilic count, yeasts-molds, lactic acid bacteria, Staphylococcusaureus, enterococci, Bacillus cereus as well as Listeria monocytogenes and Salmonellasp. Total aerobic mesophilic count was enumerated on Plate Count Agar (PCA) (LAB M) and incubation at 30°C for 48 h, yeasts-molds on Rose Bengal Chloramphenicol Agar (RBCA) (LAB M) and incubation at 25 oC for 4 d, lactic acid bacteria on de Man, Rogosa and Sharpe (MRS) (LAB M) and incubation at 30 oC for 48 h, Staphylococcusaureus on Baird-Parker Agar (BPA) (LAB M) and incubation at 37°C for 24-48 h, enterococci on Kanamycin AesculinAzide Agar (KAAA) (LAB M) and incubation at 37 oC for 24 h, Bacillus cereus on Cereus selective agar (Merck) and incubation at 30°C for 24 h, Listeria monocytogenes on Listeria Chromogenic Agar (LAB M) and incubation at 37°C for 24 h, Salmonella sp. on Xylose Lysine Deoxycholate (XLD) (LAB M) and incubation at 37 oC for 24 h. Sulphur-reducing clostridia were determined by pouring 10 mL aliquots in 20 mL of molten Sulfite Polymyxin Sulfadiazine Agar (SPSA) (Merck) and incubation at 37°C for 24 h. Finally, qualitative determination of Listeria monocytogenes and Salmonella sp. was performed according to EN ISO 11290-1 [19] and ISO 6579: 2002 [20], respectively. All analyses were performed in duplicate and the average values are presented.
2.3 Culture-dependent enumeration, isolation and identification of yeasts
From each sample, all colonies present in the final dilution were selected and isolated. Isolated strains were purified by successive subculturing on Brain Heart Infusion (BHI) agar (LAB M). Extraction and analysis of whole cell proteins by Sodium Dodecyl Sulfate – Polyacrylamide Gel Electrophoresis (SDS-PAGE) was performed according to Paramithiotis et al. [21]. Additionally, the extracted DNA [22], from the same isolates was subjected to Random Amplified Polymorphic DNA – Polymerase Chain Reaction (RAPD-PCR).
RAPD-PCR was performed in 25 μL volume containing 0.2 mMdNTPs (Peqlab, Erlangen, Germany), 2.5 mM MgCl2, 4 μΜ primer M13 (5’-GAG GGT GGC GGT TCT-3’) and 2 U Taq polymerase (Biotools, Madrid, Spain). Thermocycling conditions were as follows: initial denaturation at 95°C for 2 min; 35 cycles of 95°C for 1 min, 38°C for 1 min ramp to 72°C at 0.6°C/sec, 72oC for 2 min and a final extension step at 72°C for 10 min. DNA fragments were separated by electrophoresis in 1.5% agarose gel in 1.0x Tris- Acetic acid-EDTA (TAE) at 100V for 1.5 h, visualized by ethidium bromide staining and photographed using a GelDoc system (Bio-Rad, Hercules, CA, USA). Conversion, normalization and further analysis were performed with Bionumerics software (Applied Maths NV, Sint- Martens-Latem, Belgium) using the Dice coefficient and Unweighted Pair Group Method with Arithmetic Mean (UPGMA) cluster analysis.
Three representative strains from each cluster were subjected to sequencing of the D1/D2 region of 26S-rRNA gene according to Paramithiotis et al. [22] for species identification. PCR amplification products were sent to a commercial facility for sequencing, and the results were aligned with those in GenBank using the Basic Local Alignment Search Tool (BLAST) program to determine the closest known relatives based on the partial 26S-rRNA gene sequence.
2.4 Culture-independent assessment of microbiota
The culture independent assessment of the microbiota was performed with Denaturing Gradient Gel Electrophoresis (DGGE) as follows: fifty milliliters of the first decimal dilution were centrifuged and DNA was extracted as previously described [22]. The approximately 250 nucleotides of the 5’ end of the 26 rRNA gene were amplified by PCR in a final volume of 50 μL containing 2.0 mM MgCl2, 0.2 mMdNTPs (Biotools), 0.3 μΜ primer NL1 (5’-GCC ATA TCA ATA AGC GGA GGA AAA G-3’) with a GC clamp (5’- CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG-3’) attached to the 5’ end, 0.3 μΜ primer LS2 (5’-ATT CCC AAA CAA CTC GAC TC-3’) and 2 U Taq polymerase (Biotools). Thermocycling conditions were as follows: initial denaturation at 94°C for 3 min, 40 cycles of 94OC for 30 sec, 52°C for 30 sec, 72OC for 2 min and a final extension step at 72°C for 10 min. Separation of the PCR products took place using the Dcode Universal Mutation Detection System (Bio-Rad) with 8% (w/v) polyacrylamide gel containing urea-formamide (Applichem, Darmstadt, Germany) as denaturing agents in a concentration gradient from 30-60% in TAE buffer (40 mMTris-acetate, 2 mM Na2EDTA H2O, pH 8.5). Electrophoresis was performed at 50 V for 10 min and then 200 V for 4 hours. Then, gels were visualized by ethidium bromide staining and photographed using a GelDoc system (Bio-Rad). Species identification was performed by comigration with reference patterns.
3. Results and Discussion
Microbiological analysis of the artichoke’s bracts, before any treatment, revealed absence of lactic acid bacteria, enterococci, sulphur-reducing clostridia, Escherichia coli, Listeria monocytogenes, Salmonella sp., Staphylococcus aureus and Bacillus cereus and presence of Enterobacteriaceae at 4.30 logCFU g-1, coliforms at 3.74log CFU g-1 and yeasts-molds at 4.89 log CFU g-1. Similarly, immature inflorescencecontained,before any treatment, 2.23log CFU g-1 Enterobacteriaceae, 2.08log CFU g-1 coliforms and 4log CFU g-1 yeasts-molds. These results are in agreement with Harris [23] who stated that the microbiota of fresh vegetables is dominated by Gram negative aerobic bacteria and yeasts.
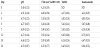
After treatment with lemon juice and blanching, all microorganisms were below enumeration limit. Immersion in lemon juice solution is a usual practice at household level when cooking artichokes. Practically, this is employed to prevent browning, which occurs mainly due to the anti-browning effect of citric acid [24]. Blanching, on the other hand, generally results in the inactivation of enzymes, softening of the structure, modification of certain mechanical properties as well as prevention of discoloration or development of unpleasant taste [25]; for some vegetables, however, (e.g. peppers, leeks, and parsley) it is recommended that blanching is avoided [26]. The time of blanching is very important as it significantly affects the loss of antioxidant properties and phenolic content in selected cruciferous vegetables [25]. However, the 1.5 min blanching that took place in the present study can be considered as minimal and, at the same time, seemed to be effective since no discoloration phenomena were observed. From a microbiological point of view, lemon juice is a common sanitizer at household level. It has been reported that 6.7% of lemon juice is the minimum bactericidal concentration against E. coli strains in tryptic soy broth [27]. On the other hand, it has been reported that blanching of whole pickling cucumbers for 15 s at 80°C reduced microbial cell counts by 2 to 3 log cycles from an initial population of typically 6log CFU g-1 [28]. Similarly, in the present study, a 4-log reduction of yeasts-molds population and a 2-log reduction of Enterobacteriaceae population were evident and can be assigned to the combined effect of lemon juice and blanching. In Table 1, total aerobic mesophilic and yeast/mould counts during fermentation, is presented. Yeasts/ moulds prevailed spontaneous fermentation of artichoke immature inflorescence reaching a final population of 5.73 log CFU mL-1 after 12 days and remained stable until the end of fermentation. All other microbial populations were below enumeration limit throughout the fermentation procedure. The initial pH and TTA values were 3.4 and 4.25, respectively. The pH value increased to 4.75 after 3 days and remained between 4.44–4.90 until the end of fermentation (Table 1). Similarly, TTA value gradually increased to 5.9 after 7 days and remained stable until the end of fermentation (Table1). The acidic initial pH and increased TTA values of the brine can be assigned to the initial processing steps of the artichoke immature inflorescence, i.e. the immersion into brine solution containing lemon juice and blanching. During these steps citric acid has penetrated the tissue and then released into the brine, affecting both pH and TTA values. Increase of the pH and TTA values during the first three and seven days of fermentation, respectively, can be assigned both to physiological phenomena related to the artichoke immature inflorescence and to the yeast metabolism. More accurately, damage of the membrane integrity during initial processing can be held responsible for release of plant cellular material into the brine. Part of this, such as ammonium ions and pectins, may act as proton scavengers that lead to increase of the pH value and part of this, such as carboxyl compounds, may result in the increase of TTA. In the former case, pH equilibrium has reached after three days at 4.75, the pK value of the second carboxyl group of citric acid and of acetic acid, a metabolite produced by the yeasts. Equilibrium has reached at this value due to the availability of both acids. Since TTA increase coincided with the increase of the yeast population, it may be primarily assigned to the yeast metabolism and secondarily to compounds released from the immature artichoke inflorescence due to the action of the brine, despite the negative effect of the proton scavenging on TTA value.
Α total of 145 colonies were isolated throughout the fermentation process and subjected to SDS-PAGE and RAPD-PCR for their differentiation; identical whole-cell protein patterns as well as genotypic profiles, as depicted by RAPD-PCR, were observed. The dendrogram obtained after the cumulative analysis of the different patterns, revealed a remarkable uniformity among them (Figure 1). Both techniques have been effectively used in clustering and differentiation between a variety of yeasts [21,29]. The resemblance between both whole-cell protein and genotypic patterns suggests, at least, a very close relationship between the isolates. Three representative isolates (namely A55, A71 and A126) were randomly selected and subjected to sequencing of the 26S rRNA gene for their identification. Alignment with the respective sequences of identified yeasts in the GenBank database revealed that the closest relatives of all three sequences belonged to Hanseniasporauvarum species; more accurately, the closest relatives of A55, A71 and A126 were sequences JX423559, HM854034 and JQ678687, respectively. However, since H. uvarum, H. meyeri, H. clermontiae, H. lachancei, H. opuntiae and H. guilliermondii are closely related species, analysis of the D1/D2 region of 26S-rRNA gene sequences was not adequate for reliable differentiation between them [30]. Therefore, the strains isolated from the present study are referred to as Hanseniaspora sp.

Application of the culture-independent approach verified the above result, at least as far as uniformity was concerned.Presence of a single band in DGGE profile, in both fermentation jars, throughout the fermentation process was evident,whereas only in the first lane, i.e. the first fermentation jar in the third day of fermentation; a second band was present (Figure 2). In the first case, comigration with strains A55 and A71 verified presence of the Hanseniaspora genus throughout the fermentation procedure. Regarding the second band present in the third day of fermentation, it was considered of minor importance and therefore identification was not performed. PCRDGGE has been successfully used for the assessment of yeast microcommunity structure and dynamics in raw milk [31], fermented meat [32] and grape must fermentation [33]. Application of both culture-dependent and independent approaches takes place when an integrated assessment of a microbial ecosystem is the case. In the present study, dominance of Hanseniaspora sp. throughout the spontaneous fermentation was suggested by both approaches. Since the same domain of 26S-rRNA gene was used for sequencing and PCR-DGGE analyses, the latter was not able to differentiate between the closely related Hanseniaspora species.

The genus Hanseniaspora has been reported to participate in the microbial community of a variety of spontaneously fermented products such as black table olives [34], cider [35], cocoa bean [36], strawberry tree fruit [37], lafun[38] and grape must [39]. Moreover, in the latter case, dominance of H. uvarum during the initial stages is very often reported as well as persistence throughout fermentation [40].
Competing Interests
The authors declare that they have no competing interests.