


1. Introduction
Left ventricular noncompaction (LVNC) is a relatively newly recognized cardiomyopathy [1,2], with morphological traits of excessive trabeculations and deep recesses [2,3]. The etiology of this disease remains controversial. While the American Heart Association classified this disease as a primary genetic disease [4] , the European Society of Cardiology classified LVNC as an unclassified cardiomyopathy [5] . In terms of clinical features, this disorder also varies widely from asymptomatic to severe ventricular dysfunction and life-threatening arrhythmia.
We report an interesting sporadic case with LVNC, in which the patient was diagnosed before symptoms emerged, experienced deterioration during adolescence, and eventually underwent heart transplantation.
2. Case Report
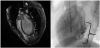
The patient, a 23-year-old man, was referred to the previous hospital at 14 years old after an abnormality was identified on an electrocardiogram (ECG) in a school-based ECG screening during junior high school. He showed no symptoms at that time. However, gallop rhythm was audible, and ECG revealed ST-T changes and an abnormal Q wave on the first visit to the hospital (Figure 1a). Serum level of brain natriuretic peptide was elevated to 1012 pg/ml. Chest X-ray showed cardiomegaly and lung congestion (Figure 1b). While no structural abnormalities were found other than persistent left superior vena cava, reduced left ventricular contraction (8.5% on left ventricular fractional shortening (LVFS)) and a dilated left ventricle (left ventricular end-diastolic dimension (LVDd), 66.8 mm) were found on echocardiography. Furthermore, deep recesses (ratio of noncompacted layer/compacted layer (N/C ratio), 2.1) were found in the posterior LV wall and blood flow was detected in the recess area on echocardiogram (Figure 1c and Figure 1d). These findings were confirmed on both magnetic resonance imaging (MRI) and LV angiography (Figure 2a and Figure 2b). LV dysfunction associated with LVNC was diagnosed. No genetic abnormalities were detected. His family history was also negative. He was admitted to the previous hospital, and therapy for chronic heart failure was initiated with furosemide, spironolactone, digoxin, enalapril and carvedilol. After gradual dose titration of these drugs, LV function gradually improved.

However, 2 years later, drug compliance was getting worse after he graduated from junior high school and LV function started to deteriorate again. He needed to be hospitalized several times to control heart failure (Figure 3). Non-sustained ventricular tachycardia (NSVT) was detected for the first time at 16 years old, and amiodarone was added.

Shortly after the second discharge from our hospital, he experienced severe palpitations followed by syncope at home. He suffered from critical VT (Figure 4) and was transported by ambulance with performance of cardiopulmonary resuscitation (CPR). Immediately after arrival to the emergency hospital, electrical cardioversion was performed. He recovered to sinus rhythm without any neurological abnormalities.

After controlling his heart failure condition and arrhythmia, an electrophysiological study and catheter ablation were performed. We identified the origin of VT on the posterolateral wall of the LV, which was coincident with the noncompacted area (Figure 5). Catheter ablation was performed on the target area. Ventricular tachycardia was not induced immediately after ablation. However, VT recurred shortly thereafter. A cardiac resynchronization therapy with defibrillator (CRT-D) device was therefore implanted. However, 3 months after CRT-D device implantation and a second session of catheter ablation, VT storm occurred. He was subsequently dependent on intravenous administration of beta-blockers. He was attached to a left ventricular assist device at 18 years old. Furthermore, heart transplantation was performed at 20 years old. Health condition has remained stable after heart transplantation.

3. Discussion
Early in the embryonic stage, trabeculation emerges in the luminal layers of the ventricles. Subsequently, the trabecular layer becomes solidified in the deeper part to increase the compact component of the ventricular myocardium. LVNC has been considered a disorder of this process [6] . Jenni et al. [3] reported diagnostic criteria an NC/C ratio >2 on 2-dimensional echocardiography and deep recesses confirmed by color Doppler echocardiography. Several causal genes have been reported for this disease [7-17]. The American Heart Association classified this disease as a primary genetic disease in 2006 [4] . Conversely, Stöllberger et al. [18] reported that LVNC was frequently associated with other cardiac/extracardiac disorders. The European Society of Cardiology classified this pathology as an unclassified cardiomyopathy in 2008 [5] . Clinical outcomes of this disease also range widely from no symptoms to life-threatening arrhythmia, cardiac failure, thromboembolism, and sudden cardiac death. Frequency of heart transplantation or death for childhoodonset LVNC has ranged widely from 5% to 52% in previous reports [2,19-26]. Brescia et al. [24] reported that cardiac dysfunction, ECG abnormalities/arrhythmia, and infant onset were associated with such mortality. Wang et al. [25] investigated the long-term prognosis of pediatric patients with LVNC in Japan. They found that risk factors for pediatric LVNC were heart failure at diagnosis and a thinner noncompacted layer. Recently, van Waning et al. [26] reported that risk factors for major adverse cardiac events in pediatric LVNC were MYBPC3 complex and MYH7 gene abnormalities, as well as LV dysfunction and infant onset.
In the current case, we successfully diagnosed a sporadic pediatric case with LVNC before symptoms emerged. The underlying LV dysfunction initially improved with therapy for chronic heart failure. This was a sporadic, juvenile, asymptomatic case, and would have been regarded as low risk in the study by van Waning [26] . However, underlying LV dysfunction was classified as high-risk group for major adverse cardiac events in the previous reports [21,23,24-26]. Furthermore, this disease is highly associated with major adverse cardiac events, including sudden cardiac death. Thus, early detection of the disease with the Japanese school-based ECG screening system is beneficial.
This case also showed the importance of education and guidance to allow adolescent patients to understand their own condition. The patient had never experienced any serious discomfort or palpitations until frequent occurrence of NS-VT developed at 17 years old. During junior high school, his parents took care of his health condition and medical therapies. We repeatedly provided information and education to the patient and his parents regarding the clinical conditions and the importance of treatment. However, understanding the importance of medical therapies and heart failure control evidently proved difficult for the patient. On the other hand, adequate recognition of the disease by his parents leads to their quick response with CPR at the first critical VT attack. Whether the deterioration of heart failure and occurrence of VT were due to drug non-compliance or the natural history of this disease remains unclear, but education of adolescent patients to understand their own pathological condition and the importance of treatment is important. These underlying issues of education for patients with transitional care are similar to those encountered in the field of adult congenital heart disease [27] .
Life-threatening arrhythmia is one of the most serious adverse events of LVNC. Increased fibrous and elastic tissue [2,28], reduced myocardial perfusion [29] , and coronary microcirculatory dysfunction [30] have been reported in the noncompacted area. Myocardial fibrosis and ischemia in the noncompacted area may be predisposing risk factors for life-threatening arrhythmia.
Muser et al. [31] reported that, in patients with LVNC and VT, the substrates of ventricular arrhythmia were coincident with noncompacted myocardial segments. On the other hand, Sohns et al. [32] reported that arrhythmic substrate differed from the noncompacted area. Sánchez Muñoz et al. [33] reported that the arrhythmic substrate in LVNC was heterogeneous. In this current case, the origin of VT was coincident with the noncompacted area. Our data were in line with a previous report by Muser [31] . Several investigators [31-33] have recently reported successful results of endocardial and epicardial catheter ablation for ventricular arrhythmia associated with LVNC. However, some LVNC patients with LV dysfunction have shown recurrent VT, heart transplantation and death after catheter ablation/implantable cardioverter-defibrillator implantation [32,33]. Sánchez Muñoz et al. reported that while only 1 in 7 LVNC patients with normal ventricular function experienced recurrent VT after catheter ablation, 4 of 11 LVNC patients with LV dilatation died or received heart transplantation. In the current case, VT was not induced after catheter ablation. However, VT subsequently recurred. Wan et al. [34] reported that late gadolinium enhancement (LGE) on MRI was observed in 40% of LVNC patients, and the distribution of LGE extended beyond the noncompacted area. Their data showed that myocardial fibrosis may spread far beyond the noncompacted area. LV remodeling associated with LV dysfunction may relate to recurrent VT.
4. Conclusion
We encountered a patient with LVNC who was diagnosed before symptoms emerged. LV function improved transiently, then gradually deteriorated. He eventually received a heart transplant due to recurrent VT and LV dysfunction.
LVNC is a relatively rare and heterogeneous disease. Each case should be treated with care and followed-up.
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
HK contributed to the conception and design of the study and the drafting of the manuscript. NO, KK, YO, Atsuko A contributed to the acquisition of data and the editing of the manuscript. Akira A contributed to the editing of the manuscript.
Acknowledgments
We greatly appreciate the assistance of Dr. Yasumasa Tsukamoto from the Department of Cardiovascular Medicine at Osaka University Graduate School of Medicine in providing information after patient transfer from our hospital.