


1. Introduction
Congenital hypopituitarism is a clinical syndrome of deficiency in pituitary hormones production. Panhypopituitarism refers to involvement of more than one pituitary hormone while involvement of one hormone refers to partial hypopituitarism It is an uncommon disorder of the hypophyseal system but could be life threatening, however, it is treatable if the diagnosis is made early [1-4].
Congenital hypopituitarism is associated with possible serious complications and long-term neurological sequelae, Neonates with congenital hypopituitarism may present with or without associated developmental defects, such as ocular, midline, and genital abnormalities. They may also present with nonspecific symptoms, including hypoglycemia, lethargy, apnea, hemodynamic instability, jitteriness, seizures, poor weight gain, failure to thrive, temperature instability, recurrent sepsis and neonatal cholestasis. It is typically detected shortly after birth, but it may occur several weeks after the neonatal period. The cholestatic jaundice most commonly associated with neonatal hypopituitarism manifests as conjugated hyperbilirubinemia with elevated alkaline phosphatase. The cholestasis resolves after replacement of glucocorticoids or growth hormone, suggesting a role of these hormones in biliary excretory function. Furthermore genetic mutations in transcription factors gene involved in the embryogenesis of the pituitary gland such as HESX-1, PROP -1 and PIT-1 were reported and implicated in the pathogenesis of congenital hypopituitarism [5-10].
This article reports on the clinical experience of congenital hypopituitarism from the pediatric endocrine service, King Khalid University Hospital (KKUH), Riyadh, Saudi Arabia over more than two decades, January 1990 to December 2017. KKUH is the major hospital of King Saud University and provide primary, secondary and tertiary health care service to the local population and also receives patients’ referral from all over the country.
2. Material and Methods
The medical records of patients who diagnosed to have congenital hypopituitarism (aquired causes has been excluded) were retrospectively reviewed. Data included were age, sex, clinical presentation, and results of the relevant laboratory investigations and radiological images; where Magnetic resonance imaging (MRI) scan was done. The diagnoses of congenital hypopituitarism were based on clinical suspicion supported by appropriate hormonal testing. The diagnoses of congenital growth hormone deficiency were made by performing one physical (sleep) and two biochemical tests, using sex hormone priming when indicated. The various hormonal testing to assess both the anterior and posterior pituitary gland functions, were performed following the specific protocol [11]. Unfortunately, no genetic studies were done in any of our patients.
3. Results
During the period under review, a total of 177 patients were diagnosed with possible congenital hypopituitarism. The mean age was 6.5 years range 0-18 years. Majority of patients with congenital growth hormone deficiency was diagnosed at a later age because of late referral. Seventy-five percent were having isolated hormone deficiency; Growth hormone 117 (87.9%), Gonadotrophic hormone 8 (6.0%), central hypothyroidism 5 (3.8%) and Adrenocorticotrophic hormone 3 (2.3%). In twenty- five percent of patients the diagnosis was multiple pituitary hormone deficiency (MPHD), in which septooptic dysplasia and other midline defects, constitute the majority (56.8%). Magnetic resonance imaging (MRI) results in 177 patients with congenital hypopituitarism revealed the majority (86.5%) of patients with isolated hormonal deficiency (IHD) were normal, while the majority (95.5%) of patients with multiple pituitary hormonal deficiency (MPHD) were abnormal. Diabetes Insipidus was found in association in five (2.5%) patients.
4. Discussion
Congenital hypopituitarism is a clinical syndrome characterized by deficiency of pituitary hormones production. This may result from peri-natal or birth asphyxia or congenital defects of the hypothalamus, pituitary gland or surrounding structures, of which septo-optic dysplasia and other midline defects constitute the majority. However, in rare occasions a genetic basis involving the pituitary transcription factors that regulate the formation of the gland. It could be partial, involving the deficiency of one hormone, or complete (pan) involving the deficiency of more than one hormone with estimated incidence between 1:3000 and 4000 births [1-5]. Onset can be at any time of life, it might be lethal if not diagnosed and treated early. It is reported in association with other diseases like Shaw Ashman-Diamond syndrome, hyperemia and slipped capital femoral epiphysis [12-14]. Jain et al [15] reported the adverse on the heart. Also, Brown et al [16] reported that in children with congenital hypopituitarism have an IQ that is below average when compared to the normal population and a reduced performance IQ when compared to sibling control, which may reflect abnormal brain development or could be linked to the impact of hypoglycemia or low thyroxine concentration in early life. In Saudi Arabia, there are no precise data on the prevalence of the disorder, however, there is an impression fostered by clinical experience and scant published data, that this is not that rare disease. [17-21].
The presentation is variable. The most important presenting feature, and perhaps most common feature, of congenital hypopituitarism is hypoglycemia [22]. This occurs secondary to the presence of GH deficiency with or without associated ACTH deficiency. The majority of patients were GH deficient in our series [23-26].
Another unique feature of congenital hypopituitarism is the presence at birth of a microphallus (small penis). This is a result of gonadotrophic hormone deficiency [27-28]. Noninfectious form of hepatitis developed in our series, the condition is suspected as liver enlarged with abnormal liver function tests [18,20,29-32]. The diagnosis of central hypothyroidism should not be over looked. Five (5.6%) patients, in our series were diagnosed to have isolated thyroid stimulating hormone (TSH) deficiency with low free thyroxine (FT4) [3,33,34]. Furthermore, three (1.7%) patients were diagnosed with isolated adrenocorticotrophic hormone (ACTH) deficiency at variable ages, Takagi et al described in a cohort of Japanese patients, the gradual loss of ACTH in patients carrying an LHX4 mutation [35-37].
Prenatal, during pregnancy, and birth asphyxia are important contributing factors for etiology. Nine (7.7%) patients of the isolated growth hormone deficiency (GH) group, (five-breech delivery, and four with birth asphyxia with low Apgar scores) had history suggestive of birth asphyxia [38]. Furthermore, severe midline defects, such as septo-optic dysplasia which include absence of the septum pellucidum, underdevelopment of the optic nerve, associated with variable degrees of impaired vision. Also, cleft lip and/or palate, choanal atresia, anomalies or absent vascular supply to the central nervous system, and encephaloceles. In this series, they constitute a majority [5,6,19,21,39-43], similar to previously reported [44]. Rarely, it might result in genetic mutations like transcription factors, involved in the regulation of the pituitary gland and its function [1,3,45]. Unfortunately, the service is not available to us.
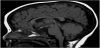
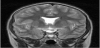
Magnetic resonance imaging (MRI) scan remains the modality of choice assessing the hypothalamic pituitary region in patient with congenital hypopituitarism. MRI scan precisely diagnose abnormality of the adenohypophysis and neurohypophysis usually the stalk, and correlate well with the clinical presentation. In a normal head magnetic resonance imaging (MRI) scan, the anterior pituitary, on T1- weighted imaging, appears dark and equal in intensity to gray matter, while the posterior pituitary gland appears white and is referred to, radiologically, as the posterior “bright spot” (Figure 1). In Septooptic dysplasia the absence of the septum pellucidum (Figure 2) is a constant finding in our series, similar to what have been reported [46-50].
5. Conclusion
Congenital hypopituitarism in not that rare in Saudi Arabia. An early diagnosis can be obtained with high accuracy based on a high clinical suspicion index. Imaging abnormalities are frequent and associated with the clinical and biochemical phenotypes. It had variable presentations, such as hypoglycemia, micro phallus in boys and neonatal cholestasis, or in association with midline deficits. There is a need to compliment hormonal and radiological investigations with gene study.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
The authors would like to thank Mr. Abdulrahman AL Jurayyan for his help in preparing the manuscript.