


1. Introduction
Primary hyperoxalurias (PH) are a group of rare autosomal recessive metabolic disorders characterized by increased excretion of oxalate and other metabolites caused by different enzymatic defect sat different steps of oxalate metabolism in the hepatocytes cytosol [1,2]. Subsequently calcium oxalate deposition in the kidneys and urinary tract takes place leading to nephrocalcinosis, stones formation and chronic kidney disease. In advanced disease when glomerular filtration rate declines, systemic oxalosis affects most vital body organs including heart, blood vessels, bones, joints and retina [3,4]. So far, 3 types of PH have been identified though more are believed to exist. PH type 1 is known to be the most severe type. It is caused by a deficiency of the enzyme alanineglyoxalate aminotransferase (AGT) associated with mutations in the AGXT gene encoded on chromosome 2 [5,6,7]. PH2 and PH3 are less common and less severe and caused by deficiency of the enzymes glyoxylate-reductase (GR) and 4-hydroxy-2-oxoglutarate aldolase 1 (HOGA1), respectively [8,9]. The phenotypic diversity of the disease in addition to its rarity leads to delayed diagnosis in many cases [10]. Epidemiology and clinical pattern of PH1 in the Middle East Region has not been reported previously. In this study we report our experience with PH1 in Kuwaiti Arab patients diagnosed in the only pediatric nephrology unit in Kuwait over a 9- year period.
2. Patients and Methods
A retrospective review of all patients with hyperoxalurias was conducted. This included all cases referred to the only pediatric nephrology center in Kuwait at Mubarak Al-Kabeer University hospital established in 1995. Demographic data of patients were analyzed including: age, gender, ethnic background, family history and consanguinity. The clinical pattern including presentation, management and prognosis were reviewed. Diagnosis of primary hyperoxaluria was suspected based on the clinical presentation of nephrocalcinosis, nephrolithiasis and/or chronic kidney disease. All patients had full blood workup including complete blood count, serum creatinine, blood urea, serum electrolytes, Sodium, Potassium, Chloride, bicarbonate level, Calcium, Magnesium, Phosphorus and alkaline phosphatase. Iron, transferrin and ferritin levels were monitored when anemia is present. Intact parathyroid hormone was checked on a monthly basis. Skeletal survey was done in some patients when indicated. All patients had full biochemical assay of plasma oxalate, 24- hour urine for oxalate, glyoxalic acid, glycolic acid (PH1)and glyceric acid (PH2) by radioimmunoassay (R.I.A) .Urinary oxalate levels were considered abnormal when values exceeded 50 mg/1.73M2/24 hours or >0.46 mmol/day as per lab values. Urinary glycolate excretion and L-glyceric acid excretion were considered abnormal when values exceeded 0.5 mmol(45mg)/1.73M2/24 hours and 5 μmol/L/day, respectively. Liver biopsy and assay of the enzyme Alanine Glyoxylate Amino-Transferase (AGT) were performed in some patients who were sent to specialized centers outside Kuwait due to lack of such facilities locally. No genetic DNA studies were performed for mutation detection in any patient. Verbal consent to be included in the study was obtained from the care givers of all patients.
3. Results
A total of 10 patients below the age of 12 years were diagnosed with hyperoxaluria between 1995-2004. Primary hyperoxaluria was reported in 9 of them (90%) while one patient had hyperoxaluria secondary to autoimmune enteropathy on total parenteral nutrition. All 9 patients with PH had type I disease. Male to female ratio was 2:1. Median age at diagnosis was 2 months (range 6 weeks-12 years). The majority of patients (66%) were infants at diagnosis, while 77% were less than 5 years of age. Mean serum creatinine level at diagnosis was 94μmol/l (range 42-268 μmol/l). Positive family history of PH1 or stone formation was reported in 6 patients (66%), while 4 patients (44%) had an affected sibling with PH1 including 2 families.
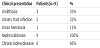
The other 2 patients with late onset of PH1 (at 9 years & 12years of age) had a strong family history of nephrolithiasis. Consanguinity with first-degree cousin marriage of parents of affected children was reported in 7 patients (77%). Table 1 summarizes the presenting signs and symptoms of patients including nephrolithiasis, urinary tract infections, gross hematuria, nephrocalcinosis and chronic kidney disease. Out of a total of 7 patients developed chronic kidney disease. five were infants at time of the diagnosis (55%). End- stage renal disease was reported in 77% of cases at a median age of 15 months (4 months -9.5 years) .Five of these patients were < 1 year of age. Only 2 patients continued to maintain normal renal function after a mean followup period of 7 months (range 5-9 months). Table 2 summarizes the clinical data of the 9 patients with PH1. Medical management of all patients included the use of pyridoxine in a dose of 10-30 mg/kg/day Bicitra, in a dose of 100-150 mg/kg/day and Magnesium Sulphate in some patients. Only one patient showed pyridoxine sensitivity with a reduction of urinary oxalate in a range of 30-50%. Gastrostomy tubes were used in 5 patients for overnight feeds giving a total fluid intake of 2-3 liters per night. Hemodialysis was initiated in all 7 patients requiring renal replacement therapy. All had 3-4 hourly sessions 4-5 times per week.

4. Outcome
At end of the study, two patients continued having normal renal function. Out of the 7 patients who were maintained on hemodialysis, 3 had combined liver- kidney transplant. Both had it in a specialized transplant centers outside Kuwait. Death was reported in 4 patients giving a mortality rate of 44%. Three of the 4 patients died while receiving renal replacement therapy due to severe multi-organ failure secondary to systemic oxalosis, pulmonary hemorrhage and septic shock, respectively. The 4th patient died in the early post-operative period following combined cadaveric liver-kidney transplantation. The patient who survived after a successful cadaveric combined liverkidney transplantation maintained normal renal function for 5 years but required a second living-related kidney graft afterwards.
5. Discussion
Primary hyperoxaluria type 1 is the most common hyperoxaluria affecting children. It accounts for 7-14% of nephrocalcinosis [11] and 2-20% of urolithiasis in children [12-13]. In Europe, PH1 was reported to be the cause of CKD in one per 5-15 million children below the age of 16 years annually [14]. In USA, at least 30 new cases/ year of ESRD are due to PH1 [15]. Reports of PH1 in the Arab world are scarce with a reported prevalence of 5.5 per million children / year among Tunisian children [16]. In Kuwait, which is a small Arab country in the Middle East, PH1 was found to be the cause of 19% of urolithiasis in children [17]. It accounts for 3.5% of CKD cases and 10.4% of ESRD cases in pediatric population in Kuwait [18,19]. The overall prevalence rate of PH1 in Kuwait was estimated to be7-10 / million child /year which are higher than rates reported in other parts of the world. This is expected in most metabolically inherited disease due to consanguinity and first degree-cousin marriages which are very popular in our geographic area.
Most of our patients (66%) were infants at diagnosis and 77% were below the age of 5 years. More than half of the patients (55%) had CKD and progressed to ESRD in infancy. This reflects the severity of PH1 in our region compared to the milder pattern of the disease reported in the Western World [20,21]. The high mortality rate of 44% in our study compared to 19% in France [20] also reflects the severity of the disease. This aggressive pattern of the disease most likely reflect the significant contribution of genetic factors in the disease manifestation. High consanguinity rate and first degree-cousin marriages are common cultural practices among Kuwaiti families particularly those with Bedouin (nomadic) roots. Most affected families came from regions in North of Kuwait where tribes of such background reside.
Genetic and DNA studies showed the association of the most common mutation of PH1, which is p.Gly 170 Arg, with a better prognosis [22]. We believe our PH1 patients do have a different type of mutation which needs to be confirmed by extensive genetic testing of the affected patients as well as their family members. Despite Kuwait’s pioneer role in Kidney transplantation in the Middle East, the lack of liver transplantation program, in addition to other cultural and religious factors, has inflicted a major financial burden on the health care system which is committed to provide the best and maximum care for PH1 patients?
6. Conclusion
PH1 is the commonest type of hyperoxalurias in pediatric population in Kuwait. The disease clearly has a more severe course compared to that in the western world evidenced by the early onset of CKD, rapid progression of the disease to ESRD in infancy and high mortality rate. This is more likely due to the strong genetic pooling caused by the high consanguinity rate which requires an extensive genetic work up.
Competing Interest
The authors declare that is no competing interests exist.
Author Contributions
Dr. Amal Al-Eisa declares that Dr. Wafa Al-Qabandi both the authors this paper contributed significantly to the design of the study and shared the care of the patients with the primary author. She also was involved in drafting and revising this paper.
Acknowledgments
We thank Mrs. Bena Rosy who helped in the secretarial work related to this manuscript.