


1. Case Report
Spinal muscular atrophy (SMA) is a common fatal autosomal recessive disorder in children characterized by progressive degeneration of motor neurons. The incidence of SMA is approximately one in 10,000 live births and the carrier frequency is as high as one in 50 [1]. The mutation rate of de novo rearrangements in SMA patients is only approximately 2% [2]. Therefore, SMA carrier screening is important, particularly when consanguineous marriage increases the risk of the disease in children.
According to the modified criteria of the International SMA Consortium, childhood-onset SMA is classified into three subtypes based on age of onset and achieved motor function [3]. SMA type 1 (SMA1) is the severe type, with onset usually before 6 months of life. SMA1 patients are never able to sit without support, and death usually occurs within 2 years of life. SMA type 2 (SMA2) is the intermediate type, with onset before 18 months of age. SMA2 patients are able to sit but unable to stand or walk unaided and death usually occurs before adulthood. SMA type 3 (SMA3) is the mild type, with onset usually after 18 months of age. SMA3 patients are able to stand and walk, and have a long life span.
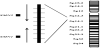
In 1990, the locus of the SMA gene was identified on chromosome 5q13 [4-6]. Then in 1995, two candidate genes for SMA were reported: the survival motor neuron (SMN) gene [7] and the apoptosisinhibitory protein (NAIP) gene [8]. There are two copies of the SMN gene: teromeric (SMN1) and duplicated centromeric (SMN2) [7] (Figure 1). These two genes differ by only two nucleotides resulting in alternative splicing at the junction of intron 6 to exon 8 [7,9]. Among SMA patients, 80-90% have homozygous deletions of the SMN1 gene [10]. In contrast, only 2-3% of carriers and controls have homozygous deletions of the SMN2 gene [10]. Most SMA cases result from mutations of the SMN1 gene. In addition, the NAIP gene, which is located adjacent to the SMN gene, is deleted in 45% of patients with SMA1 and in 18% of patients with SMA2 and SMA3 [8]. Therefore, the NAIP gene is thought to be associated with the severity of SMA.
Recently, successful prenatal diagnoses of SMA1 have been described in some reports [11-13]. Identifying a SMN1 gene mutation in the proband makes it possible to diagnose fetal SMA using amniocentesis or chorionic villous sampling. In the present report, we describe a case of prenatal gene diagnosis of SMA1.
A 34-year-old pregnant woman, 3 gravida 2 multipara, was referred to our department at 12 weeks gestation for genetic counseling. There was no family history concerning hereditary disease. Moreover, there was no consanguineous marriage in the family. The couple’s first child was died at the age of 9 months of SMA1. Genetic analysis demonstrated that the SMN1 gene was homozygously absent in the child, and both parents were identified as SMA carriers because they had heterozygous deletions of the SMN1 gene. In the second pregnancy, the fetus was prenatally diagnosed with SMA1 by chorionic villus sampling and an artificial abortion was performed. In the third pregnancy, the fetus was shownnot to be affected with SMA1 using amniocentesis, and ahealthy child was delivered (Figure 2).
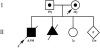
In the current pregnancy, prenatal gene analysis was performed at 16 weeks gestation using amniocentes is after genetic counseling. Real-time polymerase chain reaction (PCR) was carried out on SMN1 exons 7 and 8. The results indicated that the fetus was a SMA carrier like the parents becausethe fetus had a single SMN1 copy. A 3.310 g female baby was born via vaginal delivery at a gestational age of 38 weeks.The apgar scores at 1 and 5 minutes were 9 and 9, respectively.
2. Discussion
Since Daniels et al. performed the first prenatal diagnosis of SMA1 in 1992 [14], SMN1 gene deletions have been found in more than 98% of patients with SMA [15], and point mutations have also been reported [7]. The pathogenesis of SMA involves SMN1 gene dysfunction, and the severity of SMA is related to SMN2 gene dysfunction [16]. Accurate dosage analysis of the SMN gene copy number using a quantitative PCR assay can be used to diagnose SMA carriers and patients [17]. Presently, chorionic villus sampling or amniocentesis can be used to retrieve fetal cells for genetic testing [18,19]. In the future, genetic analysis of circulating fetal cells in the maternal peripheral blood could be applied to make clinical diagnosis of SMA [20]. Because SMA has no effective treatment, appropriate genetic counseling should be provided with genetic testing to allow families to consider the future of the fetus [21]. It is important to be aware that the carrier test may not identify all carriers, and there is a possibility of false negative results [22]. Furthermore, the risk in the next pregnancy when the two parents are carriers is 25%, with the exception of de novo rearrangements.
The reliability of prenatal diagnosis of SMA1 depends on the certainty of the clinical diagnosis in the proband [23]. If deletion of the SMN1 gene is not confirmed in the proband, then it is difficult to make a decision to undertake an invasive procedure for prenatal diagnosis.
3. Conclusion
Genetic counseling is important for couples undergoing SMA1 testing. It is particularly crucial if a couple decides to terminate a pregnancy due to the results of a genetic test because it may be difficult to digest a sense of loss. In these cases, it is also necessary to counsel parents on the risk of recurrence in a subsequent pregnancy.
Competing Interests
The authors declare that they have no competing interests exit.
Author Contributions
All the authors substantially contributed to the study conception and design as well as the acquisition and interpretation of the data and drafting the manuscript.