


1. Introduction
Multiple sclerosis (MS), referred to as “disseminated sclerosis” by the British and “sclerose en plaques” by the French, is a chronic inflammatory disease of the central nervous system with protean neurologic manifestation1. There is an increasing risk of developing multiple sclerosis with increasing latitude; but at the equator, multiple sclerosis is said to be a rarity [1,2]. A genetic predisposition is suggested by twin studies, the occasional familial incidence, and the strong association between the disease and specific HLA antigens (HLA DR2) [2]. Kurtzke defined high-, medium- and low-frequency bands of MS prevalence; high: 6-30/100000, medium: 5–29/100000, and low: 1-5/100,000 with Africa included among the low frequency areas [3,4].
2. Case Report
Neuromyelitis (NMO) is a distinct entity from multiple sclerosis and effort must be made to differentiate both entities [5]. Early studies on NMO and MS in Japan demonstrated that from both clinical and pathological standpoints there were many intermediate cases between NMO and MS5,6. Neuromyelitisoptica (NMO) is associated with the serum autoantibody NMO-IgG, which targets the water channel aquaporin-4 andmyelitis attacks in NMO are usually longitudinally extensive, spanning three or more vertebral segments on MRI [6,7]. Assay for anti-AQP4 antibodies by immunofluorescence using GFPAQP4 fusion protein-transfected HEK-293T cells revealed 83.3% and 100% sensitivity and specificity respectively of the anti-AQP4 antibody assay. Although not all patients with an NMO-like presentation will be AQP4 antibody positive, the majority are.Whereas, those with multiple sclerosis are more likely to be AQP4 antibody negative [6,7].
In the submission of the International Panel on Diagnosis of MS in May 2010, there was agreement that NMO and NMO-related disorders should be separated from typical multiple sclerosis because of different clinical course, prognosis, and underlying pathophysiology and poor responseto some available MS diseasemodifying therapies type [8]. In this communication, we present three cases to differentiate between neuro-myelitis optica and multiple sclerosisand elucidate the diagnostic challenges in communities where facilities for investigations may be limited.
2.1 Case 1
A 24-year-old female Nigerian patient was referred to the neurology clinic on account of recurrent spasm of the body of 6years duration following a febrile illness. The spasm usually started from the right lower limb then progressed to involve the upper limb and later became generalized. Associated sensory symptoms included numbness, dysesthesia, which were initially unilateral on the right but later became generalized. There was background history of progressive blurring of vision which deteriorated to complete visual loss of the left eye in the last one year and admitted to recent onset of gait unsteadiness. She had received carbamazepine for initial presumption of seizures secondary to chronic demyelinating illness which was later changed to sodium valproate on which she improved with regards to body spasms but there was continued worsening of visual impairment.
Neurological examination revealed a conscious and alert ladywith normal cognitive function, coherent speech, anisocoria, relative apparent pupillary defect on the left with no light perception on the left eye and counting finger on the right with divergent squint(nonparalytic), Bilateral pale discs were seen on fundoscopy. Nuchal rigidity was present with positive Lhermite’s sign, Motor examination showed spastic quadriparesis worse in the lower limbs, extensor plantar responseand brisk jaw jerk. Sensory examination revealed hypo-aesthesia and hypoalgesia on right side of the body. Cerebellar function was intact.
Magnetic resonance imaging (MRI) of the brain was normal and that of spinal cord revealed a T2 hyper-intensity which extended from the cervico-medullary junction to C6 (i.e. involving more than 3 cord segments). The lesion was uniformly contrast-enhancing. A single faint oligo-clonal band was noted in the CSF and not in the serum. Antibodies to aquaporin-4 was not done due unavailability. Erythrocyte sedimentation rate was 14mm/hr, full blood count revealed neutrophilic leucocytosis and retroviral screening was non–reactive. She had a 5-day pulse therapy of 1000mg of methylprednisolone with improvement.
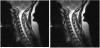
2.2 Case 2
A 33-year-old female Nigerian was first seen in the neurology service 4 years earlier with history of recurrent blurring of vision and weakness and numbness on the left side of the body. The onset was insidious, involving both limbs and eyes. The symptoms were aggravated by hot water bath and taking hot drinks (Uthotff’s phenomenon). There was a previous history of visual blurring a year before presentation which resolved completely without any form of treatment. One year later, the visual blurring recurred with associated numbness and weakness of the left side of the body which became generalized. Additional history of recurrent falls and left sided dull headaches was obtained. There was no sphincteric dysfunction.
Neurological examination revealed a conscious and alert lady; oriented, with normal cognition and coherent speech. Pupils were 3mm bilaterally and reacted briskly to light. There was no facial nerve weakness. Motor examination revealed global hyper-reflexia with ankle clonus, plantar response was equivocal, there was no cerebellar dysfunction. Romberg sign was positive. Vibration sensation was reduced up to level of anterior superior iliac spine with diminished pin prick and light touch sensation on the left side, but intact joint position sense on sensory testing.
MRI of spinal cord and brain revealed expansion from cervical segments 3 to 5 with hyper-intensity on T2 weighted image and increased signal intensity in the region of centrum semi-ovale on FLAIR sequence.Based on the above clinical findings and MRI, a diagnosis of multiple sclerosis was made. She had about 6 attacks with near total recovery on pulse therapy of methyl - prednisolone in the last 5 years with varied durations between attacks.
2.3 Case 3
A 31-year-old female Nigerian who was presented to the medical emergency on account of progressive fatigue and right upper limb weakness of five-week duration with unsteady gait and blurring of vision of two-week duration.Weakness of the right upper limb was characterized by loss of dexterity and occasional dropping of objects. It was preceded by occasional tingling and numbness in both hands and feet. Gait was unsteady and she was noticed to tilt towards the right side and associated dizziness on getting up from a seated position. There was visual blurring with associated sensation of oscillating objects at extremes of gaze. There was no headache, vomiting, fever or history of head trauma. She was neither hypertensive nor diabetic.
Neurological examination revealed a conscious and alert lady; oriented, with normal cognition and coherent speech. Relative apparent pupillary defect on the left pupil with cranial nerve VI palsy bilaterally. Lhermitte sign was positive. There was subtle facial nerve weakness on the left with other cranial nerves intact. Fundoscopy showed a pale optic disc on the left with preserved margins. Motor examination revealed increased tone and reflexes in the right upper limb with aflat plantar response bilaterally. There was no cerebellar dysfunction. Romberg sign was positive. Vibration sensation was reduced on the right up to the shoulder and on the left up to the anterior superior iliac spine but intact joint position sense.
CSF analysis showed protein of 0.41g/dl, absent cells, pronounced oligoclonal band which was absent in the serum and absent aquaporin-4 antibody. MRI of spinal cord and brain revealed a T2 hyper-intensity which extended from the C1 to C3 and multiple periventricular hyper-intensities and in the region of centrum semiovale on FLAIR sequence. Based on the above clinical findings and MRI, a diagnosis of multiple sclerosis was made.
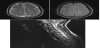
3. Discussion
The relationship between NMO and MS has long been a matter of debate. The diagnosis of NMO was limited to fulminant cases of MS. However, the discovery of an NMO-immunoglobulin G/aquaporin 4 (AQP4) antibody, has dramatically advanced our understanding of the disease, while the clinical, magnetic resonance imaging (MRI), optical coherence tomography, and laboratory examinations have clarified unique features of NMO that are distinct from MS [9]. NMO, a disease prevalent among the female gender and peak age of approximately 40 years, has been reported to be found more often among typical MS rare population, notably black Africans and it seems to be very closely related to the ‘Asian optico-spinal form of MS [3]. Early studies on MS suggested a trend with latitude with increasing prevalence in more temperate climates in Northern and Southern hemispheres [12]. The uniqueness here is that both NMO and MS are being described in latitudes where multiple sclerosis is a rarity.
In sub-Saharan Africa, where healthcare financing is poor, clinical symptoms are important in making diagnosis. However, clinical symptoms cannot reliably differentiate NMO and MS with most clinicians labelling most, if not all, cases of CNS demyelinating disease NMO.Bilateral optic neuritis is a rarity in MS but NMO may also occur as a unilateral event albeit more severe. All patients presented with unilateral visual symptoms though first case with imaging features suggestive of NMO had bilateral pale disc on fundoscopy as opposed to third. Motor symptoms are similar in NMO and MS albeit spinal cord attacks are usually clinically severe in NMO. Paroxysmal tonic spasms, which are repetitive, stereotypic, painful muscle spasms related to focal cord demyelination and usually resolve with low-dose anti-convulsant therapy, are more common in NMO as seen in the first case [11]. It is now known that NMO is not a monophasic illness similar to MS. In prototypical MS, clinical relapses are generally mild to moderate in severity, and clinical recovery is usually complete or near total. This is the situation with the second case who admitted to periods of remission with near total recovery but in contrast, NMO attacks are usually severe, and patients often only partly recover. This is a better explanation in the case of the first patient than primary progressive multiple sclerosisviz-a-viz the motor complaints and MRI features.
There is a need for dissemination in space and time for diagnosis of multiple sclerosis, according to European MAGNIMS multicentre collaborative research network, which studied MRI in patients with multiple sclerosis.The group compared the 2005 Barkhof/Tintoré basedcriteria for dissemination of lesion in space (DIS) with simplified Swanton and colleagues–based criteria in 282 clinically isolated syndrome (CIS) patients. Their findings revealed that the Swantonbased DIS criteria were simpler and slightly more sensitive than the 2005 McDonald Criteria (Bakhore/Tintore based DIS criteria)for DIS, without compromising specificity and accuracy [8]. Based on MAGNIS research network, the 2010 revision panel agreed that DIS can be demonstrated with at least 1 T2 lesion in at least 2 of 4 locations considered characteristic for MS and as specified in the original McDonald criteria (juxtacortical, periventricular, infratentorial, and spinal cord), with lesions within the symptomatic region excluded in patients with brainstem or spinal cord syndromes [8].
The 2005 revision of the McDonald Criteria simplified the MRI evidence required for dissemination in time (DIT), basing it on the appearance of a new T2 lesion on a scan compared to a reference or baseline scan performed at least 30 days after the onset of the initial clinical event [13,14]. However the 2010 McDonald’s revision panel allows a new T2 lesion to establish DIT irrespective of the timing of the baseline or with single MRI [15]. This is time saving and cost effective. The imaging findings in the second patient were in keeping with multiplesclerosis due to evidence of dissemination in space and in time based on MAGNIS group adopted 2010 revision panel. On the contrary, the first case presented showed a clean brain MRI and spinal MRI revealed hyper-intensity involving more than 3 cervical cord segment.
The revised Wingerchuk Criteria for “definite” NMO, recommends the presence of optic neuritis, acute myelitis, and at least 2 of 3 supportive para-clinical assessments (a contiguous spinal cord lesion at least 3 segments in length, brain MRI at onset that is non-diagnostic for MS, or NMO-IgG seropositivity) [5,9,16]. Indeed, the first patient has evidence supporting the three parameters though NMO- IgG was not checked due to non-availability. The clean brain MRI and presence of more than three contiguous segment cervical spinal cord affectation are enough to satisfy the 2 to 3 supportive para- clinical assessment needed.
National Multiple Sclerosis Society Task Force Diagnostic Criteria for Neuromyelitis Optica (2008) requires all three major criteria fulfilled before diagnosis of NMO or at least one of the 2 of the minor criteria [5]. The major criteria include: (a) Optic neuritis in one or more eyes; (b) Transverse myelitis, clinically complete or incomplete, but associated with radiologic evidence of spinal cord lesion extending over three or more spinal segments on T2-weighted MRI images and hypointensity on T1-weighted images when obtained during acute episode of myelitis, (c) No evidence for sarcoidosis, vasculitis, clinically manifested systemic lupus erythematosus or Sjogren syndrome, or other explanation for the syndrome. Again, there are evidences supporting the first two criteria and erythrocyte sedimentation rate of 14/hr is against presence of sarcoidosis, vasculitis, systemic lupus erythematous making room for fulfilment of 3 major criteria required by National Multiple Sclerosis Society in the first patient.
Longitudinally extensive transverse myelitis (LETM) is a neurological condition characterised by a contiguous inflammatory lesion of the spinal cord which is often associated with NMO and rarely with multiple sclerosis [7]. Considering spinal MRI, detection of an longitudinal extensive spinal cord lesion on MRI is the most specific indicator of NMO, typically described as central grey matter contiguous T2 hyper-intensity extending over three or more vertebral segments of the spinal cord and often have a core of T1 hypointensity unlike in typical acute partial myelitis attacks that usually result in shorter lesions located in the cord periphery [11]. There’s a longitudinal extensive spinal cord lesion in the cervical region in the first case but lesion in the spinal MRI of second patient presented though spanning more than threesegments, it is not contiguous.
Unique CSF oligo-clonal bands, which are detectable in approximately 85% of people with multiple sclerosis, are also found in only 20% to 30% of NMO[2], making room for a single faint oligoclonal band that was noted in the CSF of the first patient.Repeated cerebrospinal fluids of patients with NMOand multiple sclerosis showed variation of oligo-clonal bands and opposite trends in the two groups, while in multiple sclerosis oligo-clonal bands were detected in 399 of 411 patients (97%) and never disappeared but in NMO, they were detected in three of 11 patients (27%) and always disappeared [17].
From a pathological point of view, based on studies on actively demyelinating lesions in a large sample of multiple sclerosis biopsy and post-mortem samples, four different patterns were developed by Lucchinetti and colleagues originally [2,11]. Unlike MS lesions, eosinophils and neutrophils are common in the inflammatory infiltrates of active NMO lesions, and penetrating spinal vessels are frequently thickened and hyalinised. Immunoglobulin and complement components are deposited in a characteristic vasculocentric rim and such lesions are similar to, but still distinct from, the pattern II pathologically defined active demyelinating lesion patterns of MS [11,16].
Therapeutic regimens are either disease specific (immunosuppressive or immunomodulatory) or symptomatic. Natalizumab is significantly more effective than conventional disease-modifying therapy– interferon β and glatiramer acetate, while mitoxantrone may be used as induction or rescue therapy in patients with rapidly progressive relapsing remitting or secondary progressive multiple sclerosis [9,16]. Studies of rituximab, and other monoclonal antibodies in relapsing remitting multiple sclerosis, primary progressive multiple sclerosis and NMO are being undertaken with promising results. In a 6 month randomized controlled trial, oral agent, fingolimod, demonstrated a reduction in relapse rate of over 50% and reduction in enhancing lesions. Despite recent advancements in therapy, availability of the aforementioned agents remains a problem in low resource settings like ours and thus we depend on steroid-based symptomatic treatments.
There have been few publications on multiple sclerosis in Nigeria, in association with pregnancy [19], locked in state [18], alcoholism [19] but none of theseprevious publicationsdescribed longitudinally extensive spinal cord lesion, recurrent spasms responding to anticonvulsant or Uthtoff phenomenon. It brings to fore a possible question, “are we missing multiple sclerosis?”

Competing Interests
The authors declare that they have no competing interests.