


1. Introduction
Neonatal seizures (NS) constitute the most frequent and distinctive neurological symptom in the neonatal period. The incidence is estimated to be between 1.5 and 5.5/1000 living births, whose onset being during the first week in 80% of cases [1]. Neonates may present with different types of seizures: clonic, tonic, myoclonic (axial, focal, erratic), epileptic spasms, and subtle seizures, including autonomic signs or automatisms [2]. Seizures in the neonatal period differ considerably from those observed later in life with respect to their aetiological profile and clinical presentation [2,3]. NS often represent the first clinical indicator of a central nervous system (CNS) dysfunction. Although 40-50% of them are secondary to hypoxicischemic encephalopathy (HIE), other less frequent etiologies must be taken into account in the diagnostic work-up including infections, cortical malformations (readily identifiable through routine testing and imaging) and inborn errors of metabolism (IEMs) [3,4]. Diagnosis can be quite difficult, thus, a high index of suspicion is required [4]. Notably, when epilepsy occurs in a patient with IEM is commonly associated to other neurological and extra-neurological symptoms , that may address appropriate laboratory and neuroimaging investigations [5]. Epilepsy occurring in newborn with IEM may be classified according to clinical or etiopathogenetical criteria. From a pathogenetic point of view they can be divided into epilepsies due to 1) “intoxication-type” disorders of intermediary metabolism; 2) neurotransmitters defects and related disorders; 3) disorders of energy metabolism; 4) storage disorders with impaired neuronal function and 5) IEMs associated to brain malformations [6,7]. The identification of a treatable disorders is always mandatory in IEMs.
This review emphasizes the importance of considering an IEM in the differential diagnosis of neonatal seizures, discusses red flags for metabolic origin of seizures, and provides an overview of diagnosis and treatment.
2. Overview of Clinical Manifestations
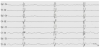
Pre-existing abnormal neurological exam and state of behavior in a newborn with appearance of seizures may arise suspicion of an underlying metabolic disorder [6]. Abnormal tone and hypo/ areflexia, lethargy, poor sucking, muscular hypotonia, irritability and apnea may be evident although they can be found in more common acute neonatal disorders like sepsis, severe heart insufficiency or HIE [3]. The co-occurrence of metabolic acidosis, hypoglycemia, cardiac disorders and liver disease may be supportive features [5,6]. Seizures are mostly recurrent or configure status epilepticus lasting for hours to days, refractory to antiepileptic treatment [7]. An IEM should be suspected in a neonate presenting with seizures, particularly if unexplained and refractory [6]. Moreover, several metabolic disorders present with specific epileptic encephalopathies associated to quite suggestive EEG patterns [8]. Early Myoclonic Encephalopathy (EME) is one of the most severe, drug-resistant epilepsy with poor prognosis with onset within the neonatal age mostly occurring in newborns with IEMs. Patients present marked hypotonia or hypertonia, opisthotonus, apneic spells and abnormal eye movements [2]. The main epileptic triad encompasses erratic or fragmentary myoclonus, simple partial seizures and tonic spasms [2]. Bursts of paroxysmal activity alternating with lack of activity define the suppressionburst (SB). EEG pattern that is a striking feature of EME (figure 1). Several IEMs may be associated to EME such as non-ketotic hyperglycinaemia (NKH), propionic or methylmalonic acidurias, methylene tetrahydrofolate reductase deficiency, GABA transaminase deficiency, serine deficiency, congenital glutamine deficiency, sulfite and xantine oxidase deficiency, and vitamin- responsive syndromes as pyridoxine, pyridoxal-phosphate, folinic and biotin deficiencies [2]. Other less specific types of seizures such as focal or generalised clonic, tonic and/or myoclonic ones may frequently occur [8].
3. Intoxication Type Disorders
3.1 Urea cycle defects
The urea cycle disorders (UCDs) constitute the final common pathway for the excretion of waste nitrogen caused by single genes defects of each of the enzymes involved into ammonia detoxification [9]. The components of the pathway are: carbamyl phosphate synthase I (CPSI); ornithine transcarbamylase (OTC); argininosuccinic acid synthetase (ASS); argininosuccinic acid lyase (ASL); arginase (ARG) and the cofactor, N-acetyl glutamate synthetase (NAGS) [6]. Deficiencies of CPSI, ASS, ASL, NAGS, and ARG are inherited in an autosomal recessive manner. OTC deficiency is inherited in an X-linked manner [9]. Infants with a UCD often appear normal initially but rapidly develop cerebral edema and the related signs of lethargy, anorexia, hyperventilation or hypoventilation, hypothermia, seizures, neurologic posturing, and coma [10]. In milder (or partial) UCD, ammonia accumulation may be triggered by illness or stress at almost any time of life, resulting in multiple mild elevations of plasma ammonia concentration; the hyperammonemia is less severe and the symptoms more subtle [6]. Seizures are frequent during the early stages of hyperammonaemia, expecially in newborns [8]. The EEG may shows variable pattern of epileptic discharge, i.e., multifocal spike- and sharp-wave discharges, repetitive paroxysmal activity, unusually low-voltage fast activity, and findings consistent with complex partial seizures [7]. The therapy of UCD include dialysis to reduce plasma ammonia concentration, intravenous administration of arginine chloride and nitrogen scavenger drugs to allow alternative pathway excretion of excess nitrogen, restriction of protein for 24– 48 h to reduce the amount of nitrogen in the diet, reduce catabolism [6].
3.2 Organic Acidemias
3.2.1 Maple syrup urine disease
Maple syrup urine disease is the prototype of the disorders of catabolism of branched-chain amino acids (BCAA; leucine, isoleucine, and valine) and is caused by deficiency of the branchedchain keto-acid (BCKA) dehydrogenase enzyme [11]. Neonatal presentation with poor feeding, vomiting, lethargy and abnormal movements (rhythmic boxing and cycling movements of the limbs), fluctuating ophthalmoplegia, and seizures is quite common [8]. Early detection is critical, as initiation of therapy within the first 5 days may be associated with near normal cognitive outcome [12]. The diagnosis is suggested by the odor of maple syrup or burnt sugar in cerumen at 24 to 48 hours of life or in urine during the latter part of the first week and is confirmed by detecting increased values of BCAAs and BCKAs in blood and urine (2,8). MRI scan may show brain edema affecting the myelinated white matter (cerebellar white matter, dorsal brainstem, cerebral peduncles, posterior limb of the internal capsule, and peri- Rolandic cerebral white matter), thalami, and globi pallidi [5]. Vasogenic edema due to blood–brain barrier disruption may occur in MSUD mainly related to water increase in the extracellular spaces and mostly evident during acute metabolic decompensation [13].
3.2.2 Isovaleric acidemia
Isovaleric acidemia (IVA) is due to a defect of isovaleric acid CoA dehydrogenase gene with increased plasma and urine levels of free isovaleric acid, 3-hydroxyisovaleric acid, N-isovalerylglycine and isovalerylcarnitine [6]. Diagnosis can be strongly suggested by the typical “sweaty feet” odor of the patients’urines [6]. Seizures may occur within the course of acute metabolic decompensation or as a consequence of intracranial hemorrhages (subarachnoid, intra- or periventricular, cerebellar, and diffuse, petechial lesions in the white matter) that have been reported in IVA [5]. Hemorrhages may be the result of various factors such as CNS edema due to accumulation of abnormal organic acids, thrombocytopenia, coagulopathy secondary to associated liver disease, and as a complication of anticoagulation therapy during hemofiltration [5].
3.2.3 Propionic acidemia
Propionic acidemia presents during the neonatal period with vomiting, dehydration and rapid deterioration, after a short symptomfree interval [14]. Seizures may present as focal or generalised, spasms and myoclonic jerks [8]. Diffuse swelling may be present on MRI of neonatal onset PPA [15]. Basal ganglia may be normal in PPA during neonatal life, whereas, lesions of the globi pallidi and delayed myelination are typically identified during later ages. An increased frequency of intracranial hemorrhages has been also reported in PPA [15,16].
3.2.4 Methylmalonic acidemia
Methylmalonic acidemia presents in the neonatal form with rapid deterioration after a short symptom-free interval [17]. Seizures may complicate acute metabolic decompensation. Diffuse swelling may be present on MRI of neonatal onset MMA [5]. Imaging in neonatal MMA may also show swelling, while later imaging reveals volume loss, delay in myelin maturation, calcification of the basal ganglia, and focal necrosis of the globi pallidi [5]. Additionally, an increased frequency of intracranial hemorrhages has also been reported in MMA [15].
3.2.5 Glutaric aciduria type 1
Glutaric aciduria type 1 (GA1) is an inborn error of lysine, hydroxylysine, and tryptophan catabolism [18]. Typically, GA1 presents in later infancy as an acute encephalopathy with predominant dystonia and dyskinesia due to necrosis of the basal ganglia, particularly affecting the putamina [19]. However, neonates may present with macrocephaly and subtle neurological signs such as hypotonia, irritability and jitteriness. Seizures usually occurr within the context of acute decompensation in association with symptoms of rapid deterioration. Dyskinetic movements may be often misdiagnosed for seizures [20]. Urine organic acids profile shows increased 3-OH- glutaric acid and glytaryl carnitine as the major peak [6]. Neuroimaging typically shows enlarged frontotemporal CSF spaces, wide Sylvian fissures, and a large cavum septi pellucidi [5]. Treatment relies on dietary restriction, carnitine and vitamin supplementation [6].
4. Neurotransmitters Defects and Related Disorders
4.1 Vitamin responsive seizures
4.1.1 Pyridoxine or pyridoxal-phosphate-dependent seizures
Pyridoxine or pyridoxal-phosphate responsive epilepsies must be considered in a neonate with unexplained and refractory seizures, with onset before or shortly after birth [20]. Seizures are mainly prolonged or recurrent, configuring status epilepticus or EME [6]. Rarely they appear to be brief and intermittent [8]. They constitute a group of metabolic disorders that share as common mechanism a defective production of pyridoxal phosphate (PLP), the active form of pyridoxine. The first one, pyridoxine-responsive epilepsy, is caused by mutation in the ALDH7A1 gene encoding for the protein antiquitin, involved into lysine catabolism within CNS [21]. Antiquitin deficiency results in increased alpha-aminoadipic semialdhyde (alpha-AASA) and piperideine-6-carboxylic acid (P6C) [21,22]. P6C has been shown to inactivate PLP leading to a secondary deficiency [21,22]. The final dysfunctional pathway is brain GABA deficiency leading to an imbalance between excitatory and inhibitory activity and reduced epileptic threshold. Intravenous administration of pyridoxine (100 mg) induces cessation of clinical seizures and electrographic discharges within minutes, however, seizures may relapse and pyridoxine may be repeated up to 500 mg total within 24 hours or continued at 30/mg/kg for seven days in case of partial response [6]. Alternatively, in patients with persistence of seizures 30- 40 mg/Kg pyridoxal phosphate may be attempted [6]. Pyridoxamine 5’-Phosphate oxidase deficiency causes PLP dependent seizures by means of an impaired dietary vitamin B6 conversion into PLP. The first patients reported were preterm with neonatal encephalopathy, early acidosis and hypoglycemia [21]. The PLP dependent enzyme activity, aromatic L- aminoacidic decarboxylase, is reduced with consequent low CSF concentration of the dopamine and serotonin catabolites, homovanillic acid and 5-hydroxyindoleacetic acid, and high L-DOPA catabolites [6]. Chronic therapy with oral PLP 30-50 mg/kg/d may induce seizures recovery. Folinic-responsive seizures were reported in few newborns with encephalopathy and apneas within 5 days after birth, ceasing with 3-5 mg/kg/d enterally of folinic acid. These patients were shown to have mutations in ALDH7A1 gene, thus, the administration of adequate doses of pyridoxine was proposed [23].
4.1.2 Biotin-responsive disorders
Biotinidase deficiency is an autosomal recessive disorder characterized by reduced production of biotin both from esogenous and endogenous sources, resulting in reduced or absent biotin in plasma and urine [6]. Biotinidase residual activity is low or absent in serum and diagnosis is confirmed by molecular analysis [6]. Symptoms may appear also during neonatal age and include hypotonia, lethargy, respiratory abnormalities and skin lesions that are the hallmarks of the disease [24]. Patients may present with eczematoid dermatitis covering large parts of the body and/or alopecia [25]. Seizures may occur mostly with generalized tonic-clonic or myoclonic features. Neonates or infants may benefit from biotin supplementation 5-10 mg/d, in contrast, untreated older patients may develop irreversible brain damages [2].
Patients with holocarboxylase synthase deficiency show acuteonset of lethargy, hypothonia, vomiting, hypothermia and seizures within the first days of life. Severe metabolic acidosis, ketosis and hyperammoniemia may lead to misdiagnosis with classical organic acidurias [26]. It is an autosomal recessive disorders due to mutations of HCS gene on 21q22.1. Diagnosis is allowed by detection of hyperammoniemia, high plasma and CSF lactate, organic acids in urine and CSF, and, confirmed by molecular analysis [6]. Treatment is based on oral biotin administration 5-10mg/d [6].
4.2 Non-ketotic hyperglycinaemia
Non-ketotic hyperglycinaemia (NKH) is a rare IEM manifesting with severe drug-resistant epilepsy and neonatal encephalopathy [27]. The more common neonatal type is a severe glycinergic encephalopathy occurring a few hours after birth characterized by lethargy, hypotonia, apneic attacks, hiccup and weak Moro response leading to deep coma without biochemical evidence of ketoacidosis [28]. EME is a striking feature of NKH and a typical suppression-burst (SB) pattern is commonly observed at the EEG record. Response to antiepileptic drugs is poor [2]. The biochemical bases lay on changes in one of the four proteins that compose the large enzyme complex involved into the glycine cleavage system (GCS), but there is no phenotype-genotype correlation. The mechanism underlying the SB pattern partly depends on glutamate - or glycine, the coneurotransmitter for NMDA transmission - overflow, mainly in the immature brain [29]. Although glycine encephalopathy has a very severe outcome in its classical expression, it may be transient in the neonatal period, for reasons yet not identified [8]. Diagnosis is supported by determination of high plasma glycine levels in absence of biochemical markers of an organic acidemia (mainly propionic and methylmalonic acidemias) with a simultaneously elevated CSF glycine [6]. Magnetic resonance Imaging (MRI) detected in some of these patients brain malformations such as dysgenesis of corpus callosum and gyral abnormalities [5].
Treatment may advantage of dietary glycine and serine restriction. Administration of sodium benzoate, dextromethorphan or pantothenic acid did not induce clear benefits in previous reports [6].
4.2.1 Serine deficiency
Serine deficiency is a rare disorder of serine metabolism caused by deficiency of two enzymes: 3- phosphoglycerate dehydrogenase (3- PGDH) and 3-phosphoserine phosphatase ( 3-PSP) [6]. Diagnosis is possible with dosing concentrations of serine and glicine in plasma and CSF [30]. Newborns show congenital microcephaly and seizures, usually starting as generalized tonic-clonic seizures and epileptic spasms [8]. The EEG shows hyparrhytmia or multifocal epileptic discharges and poor background activity [30]. Treatment consists on oral serine administration 200-600 mg/kg/d [6].
4.2.2 Creatine deficiency
Creatine is a crucial compound for energy metabolism and is carried to the brain and muscle by a specific transporter [31]. Three inherited defects in the biosynthesis and transport of creatine were reported including : guanidinoacetate methyltransferase deficiency (GAMT gene), L-arginine-glycine amidinotransferase deficiency (GATM gene) and the X-linked creatine transporter deficiency (due to the SLC6A8 gene mutations) [31]. GAMT deficiency has been usually associated to the worst phenotype and it may be disclosed within the first month of life with epileptic seizures [31]. Diagnosis need to be supported by brain magnetic resonance spectroscopy showing reduced or completely absent creatine peak [31]. A reduced creatine peak has been shown as early as 9 days of age [5]. Creatine supplementation may partially restore brain creatine levels and provide clinical improvement [6].
4.2.3 Disorders of GABA metabolism
GABA-transaminase deficiency and succinic semialdehyde dehydrogenase deficiency (SSADH) are two inborn errors of GABA metabolism [6]. GABA-transaminase deficiency is a very rare disease with only few reported cases possibly associated to neonatal-onset seizures [32]. It is later characterized by abnormal development, epilepsy with high levels of GABA in serum and cerebrospinal fluid [32].
5. Defects of Energy Metabolism
Mitochondrial disorders can be the underlying cause of neonatal refractory seizures [33]. To date, they are defined as that group of disorders due to defects of the respiratory chain complexes [34]. Epilepsy has been associated to 26-60% of all mitochondrial disorders, however, few of them show neonatal onset [33,34].
5.1 Pyruvate dehydrogenase complex
Pyruvate dehydrogenase complex (PDHc) is a multienzyme that catalyzes the conversion of pyruvate to acetyl-CoA with subsequent oxidation and entering into the Krebs’ cycle [6]. PDHc subunits are encoded by nuclear genes and inherited in a autosomal recessive manner. Only the E1α-subunit gene is located on chromosome Xp22.3 and is mostly severely expressed in males. PDHc deficiency is an important cause of neonatal encephalopathy associated with lactic acidosis [6]. Clinical symptoms include severe muscular hypotonia, lethargy, poor sucking, microcephaly, facial dysmorphic signs, and tachypnea [33]. Epileptic seizures occur in about one-third of the patients. Diagnosis relies on detection of markedly increased level of lactate in blood and CSF or only in CSF is mandatory for the diagnosis [6]. MRI scan disclose severe cortical/subcortical atrophy, dilated ventricles and brain malformations including complete corpus callosum agenesis, pachygyria or heterotopias [5].
5.2 Mitochondrial oxidative phosphorylation disorders
Oxidative phosphorylation (OXPHOS) disorders may present in the neonatal period [6, 33, 34]. The OXPHOS system comprises the mitochondrial respiratory chain complexes (complexes I–IV) and adenosine triphosphatase (complex V) [34]. In newborns, deficiency of complex I, II, and IV have been reported [34]. Features of OXPHOS disorders at birth and before include fetal hydrops, IUGR, prematurity, respiratory disturbances, poor feeding, vomiting, lactic acidosis, and lack of a symptom free interval following birth [6,33]. Newborns with neurologic and behavioral deterioration, lethargy and seizures that present with marked lactic acidosis in blood and CSF are suspected to have OXPHOS deficiency [34]. MRI in newborns may be less indicative than infants that usually shows Leigh syndrome features. Common findings include cerebral atrophy, white- matter abnormalities, involvement of the posterior columns in the lower brainstem, pontine corticospinal tracts and subcortical white matter, and an HIE-like involvement of the cortex and thalami in the absence of obstetrical history of birth asphyxia [5].
5.3 Molybdenum cofactor and sulphite oxidase deficiencies
Sulphite oxidase deficiency (SOD) can present as an isolate finding or associated with molybdenum cofactor (MOCO) deficiency and may both occur during the first days of life with seizures, feeding difficulties, and vomiting [6]. The diagnosis is based on high sulfite level in fresh urine. S-sulfocysteine and taurine concentrations are also increased. Low uric acid levels in serum and urine and increased urinary xanthine and hypoxanthine concentrations allow to distinguish MOCO deficiency from isolated SOD [6,35] . Isolated sulfite oxidase deficiency (ISOD) that may present with neonatal encephalopathy and seizures [35]. Diagnosis of ISOD may be complicated by the neuroimaging findings that may resemble those of HIE thus it is not surprisingly that most of the SOD cases are initially misdiagnosed as HIE [36]. However, differently than newborns experiencing HIE, those affected from ISOD usually do not stabilize few weeks after delivery [36].
6. Storage Disorders
6.1 Peroxisomal disorders
Peroxisomal disorders with neonatal-onset and seizures include Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD) and Refsum disease and rhizomelic chondroplasia punctate (RCDP) [8]. ZS is a autosomal recessive disorder that could manifest at birth reflecting the ubiquity of peroxisomes and is characterized by multiple congenital abnormalities involving the eyes, bone, liver, kidneys, endocrine glands. Brain malformations may cause neonatal severe hypotonia and seizures [37]. The intracellular accumulation of VLCFA damages developing organs (e.g. liver, bone, kidneys) and is especially deleterious to the organizing brain with disorganization of the physiological structure of the neocortex. The cortical abnormalities include cortical gyral abnormalities (lissencephaly, pachygyria, polymicrogyria), generalized or focal leukoencephalopathy, and brain atrophy [5]. NALD is a autosomal recessive disease characterized by accumulation of complex lipids, including cholesterol, cholesterol esters, total phospholipids, total galactolipids, and gangliosides. Clinical onset may be at birth with deafness and blindness, severe hypotonia and failure to thrive [38]. A retinopathic "leopard spot" is a pathognomonic sign. Seizures may occur during the first weeks of life as focal or generalised, often involving limbs or perioral area [5]. Interictal EEGs of the patients with ZS showed infrequent bilateral multifocal spikes, predominantly in the frontal and pre-frontal motor cortex Patients with NALD had tonic seizures or epileptic spasms drug resistant. Interictal EEGs showed high-voltage slow waves and bilateral multifocal spikes [5,6].
6.2 Krabbe Disease or Globoid-Cell Leukodystrophy
Krabbe disease or Globoid-Cell Leukodystrophy (GLD) is a neurodegenerative disorder with neonatal-infantile onset that is due to the deficiency of the lysosomal enzyme galactocerebroside β-galactosidase with subsequent accumulation in brain and other tissues [39]. Neonatal onset of GLD is exceptional [40] but clinical presentation is similar to the infantile form. Newborns show hypotonia, macrocephaly and rarely seizure [40]. At 3-6 months of age these infants show arrest of psychomotor development and framework of generalized spasticity [39]. Neonatal MRI may be normal although abnormalities of the lateral thalami, corona radiata, and dentate nuclei have been reported in one newborn [5]. Diagnosis is confirmed by detecting low or absent β-galactosidase levels in plasma, measurement of galactosylceramidase activity in leuckocytes or cultured skin fibroblasts and molecular analysis of the galactosylceramidase gene (GALC) located on 14q31 [6].
7. Conclusion
Seizures may represent a ‘red flag’ for diagnosis of an IEM in newborns. Several types of seizures or epileptic syndromes have been associated to IEMs with neonatal onset and associated to a variable phenotypic expression. Diagnosis of IEM in a newborn with seizures could be a challenge in particular when the clinical picture and even neuroimaging findings might resemble those of the more common cause of neonatal encephalopathy such as HIE or sepsis without signs and symptoms of clear metabolic decompensation. As a rule metabolic investigations should be assessed in a newborn with seizures expecially when epilepsy is not the sole neurological manifestation and they occurs together to other extraneurological signs and symptoms. Furthermore, specific adjunctive compounds supplementation or dietary restriction may play a synergic action with antiepileptic therapy.
Competing Interests
The authors declare that they have no competing interests.
Abbreviations
NS : neonatal seizures; CNS: central nervous system; IEM: inborn errors of metabolism; EEG: electroencephalogram; EME: early myoclonic encephalopathy; NKH: non-ketotic hyperglycinaemia; HIE: hypoxic-ischemic encephalopathy; UCDs: urea cycle disorders; CPSI: carbamyl phosphate synthetase I; OTC: ornithine transcarbamylase; ASS: arginine succinic acid synthetase; ASL: arginine succinic acid lyase: ARG: arginase; BCAA: branched chain amino acids; BCKA: branched chain ketoacids; MRI: magnetic resonance imaging; IVA: isovaleric acidemia; PPA: propionic acidemia; MMA: methylmalonic acidemia; GA1: glutaric acidemia I; PLP: pyridoxal phosphate; ALDH7A1: antiquitin gene; P6C: piperideine-6- carboxylic acid; 3-PGDH: 3-phosphoglyceric dehydrogenase; 3-PSP: 3- phosphoserine phosphatase; GAMT: guanidinacetate acid methyl transferase , OXPHOS: oxidative phosphorylation; PDHc: pyruvate dehydrogenase complex; SOD: superoxide dismutase; MOCO: molybdenum cofactor; ZS: Zellweger syndrome; NALD: neonatal adrenoleucodystrophy; RF: Refsum disease; GLD: globoid leucodystrophy.